Gaucher disease usually called a disorder of sphingolipid metabolism, which is a response to a deficiency of the enzyme that destroys glucocerebroside; such a complication can lead to the deposition of glucocerebroside. Symptoms of Gaucher disease most often include hepatosplenomegaly or changes in the central nervous system. In order to correctly diagnose the disease, it is necessary to conduct cytochemical studies of leukocytes.
It is a disease that is not so common; it is transmitted hereditarily when both parents are carriers of the defective gene. Gaucher's disease first appeared on the pages of medical manuals in 1882.
The lack of the enzyme beta-glucocerebrosidase in membrane-surrounded cellular organelles can lead to the formation of a large amount of nutrient medium for microorganisms of this organic substance in the cells of the macrophage system of the whole body, as a rule, this process occurs and develops in the glands, as well as in the cells of the bone marrow and spleen.
To date, science has established three types of Gaucher disease:
- Type 1 is most often found in people who have passed puberty, and is also permanent; this type cannot be characterized by the presence of neuronopathy. Type of disease number 1 can be called the most sluggish and common type, in which the central nervous system will not be affected.
- Type 2, in which children are the targets, is not so common in science. With this type of disease, as a rule, neurons are affected, which entails almost complete atrophy of the entire nervous system. With this diagnosis, the child dies while still an infant.
- Type 3 is usually called “juvenile” in science; in this type, the symptoms of the process are less pronounced, in which case atrophy of neuron cells is inevitable. It is worth noting that type 3 is also quite rare. Scientists characterize this type of disease by a gradual as well as chaotic involvement of the entire nervous system in this process.
The fact that Gaucher disease can exist in different external forms, as well as in conditions in which there is a different internal structure, confirms the diversity of changes in the highly structured glucocerebrosidase gene on chromosome 1. Despite this, diseases of varying degrees of severity can be traced among a given genotype . The main place in the question of the power of transformation is given to a sharp increase in the number of macrophages in organs and tissues, which is a response to the appearance of a large amount of glucocerebroside, however, the methods of its functioning are still not known.
The Gaucher zygote, as a rule, is like an oval and has a size of about 70-80 mm in diameter, as well as a pale cytoplasm. It contains two or more nuclei with increased pigmentation, which are shifted to the periphery. In the middle of these nuclei there are thread-like protein structures that are located simultaneously in relation to each other.
During the development of the disease, beta-glucocerebroside accumulates, which ultimately originates from disintegrated plasmalemmas, has the property of becoming a sediment in membrane-surrounded cellular organelles and forming elongated tubes measuring twenty and sometimes forty mm in length; these tubes can be seen when an increase of 2-3 thousand times. Such zygotes can be found in CML, as well as in tumors of the B-lymphocyte system, since as a result of these ailments, an accelerated process of beta-glucocerebroside metabolism is observed.
Symptoms of Gaucher disease
In the normal state, an organic substance is observed that destroys glucocerebroside, which hydrolyzes glucocerebroside, while forming glucose and ceramides. If during the development of the organism there was damage to organic matter obtained at the genetic level, then this can lead to the fact that the cells begin to capture and digest solid particles, thereby creating Gaucher zygotes. Accumulation of these zygotes in spaces
around the vessels in the substance of the human brain provokes the process of replacing dead or replaced neurons with glia cells. There are 3 types, which differ in the pattern of occurrence and spread of diseases of various etiologies, the activity of organic matter, as well as the nature of the manifestations:
Type 1 is characterized by the highest frequency of occurrence - this type is found in 90% of the population (non-neuronopathic).
The activity, which can be called residual, observed in organic matter, has the highest rate. The first manifestations can occur from 2 years to old age. The main symptoms are changes in bone cells, slow development in terms of physiology, delayed activity during puberty, and hemorrhages in the skin. The last symptom, accompanied by nasal hemorrhages, is quite common. After taking an x-ray, doctors usually find that the ends of the long bones have been widened and the bony plate of the mouth has become thinner.
Type 2 is characterized by the lowest frequency of occurrence (acute neuronopathic). With this type, a decrease in the residual activity of organic matter is observed. The first serious signs can be detected at an early age - after birth. The main symptoms are rapidly developing neurological disorders: inelasticity, unfortunately, this type in most cases leads to death at the age of about two years.
Type 3 is between the most common and the rarest (subacute neuronopathic). The vital activity of organic matter, as well as the severity of the disease, accordingly, are intermediate between types 1 and 2. The first symptoms of this type can be detected in childhood. Clinical manifestations may vary depending on the variety, and also include incoordination (Ilia), infection of organs and bone tissue (Nib) and degenerative diseases of the central nervous system with corneal opacification (Cs). If with this type the patient survives the teenage stage, in the future he, as a rule, lives for a long time.

Diagnosis of Gaucher disease
Diagnosis of this disease usually involves a cytochemical study of leukocytes. Types, as well as carriage, are usually identified based on analysis of the nature of mutations. Gaucher zygotes have diagnostic value.
VIDEO
Treatment of Gaucher disease
For types 1 and 3, replacement treatment with special complex drugs using placental or recombinant glucocerebrosidase is recommended; for type 2, treatment, unfortunately, is useless, moreover, it is completely unknown to science and medicine. During treatment, a change in the enzyme occurs for its rapid and timely transport into a membrane-surrounded cellular organelle. Patients treated with special complex medications are prescribed daily monitoring of the level of blood dye, as well as colorless blood cells; constant monitoring of the size of the liver and spleen using CT or MRI; constant monitoring of bone tissue lesions with complete observation of the skeletal system as a whole, dual-energy x-ray absorptiometry scanning or MRI.
As a rule, patients are prescribed the following drugs: Miglustat, which must be taken in certain doses, namely, three times a day, one hundred mg orally, Miglu-stat - this type of medication reduces the concentration of glucocerebroside, and also becomes a kind of solution for patients who, for certain reasons are unable to undergo treatment with enzyme replacement therapy.
It is usually prescribed to patients with anemia, as well as when the number of leukocytes and platelets in the blood decreases, as well as when the spleen increases in size, which begins to cause discomfort.
For thorough treatment of patients with this disease, doctors resort to stem cells, however, this type of treatment is the most dangerous for the health and life of the patient, therefore it is used as rarely as possible.
The disease belongs to lysosomal storage diseases (glucosylceramide lipidosis).
Characterized by deficiency of the enzyme glucocerebrosidase. This leads to metabolic disorders. Lipids are not broken down into re-consumption products, and glucocerebroside accumulates in macrophage cells. They enlarge, take on the characteristic appearance of soap bubbles and settle in the tissues of the body.
Gaucher syndrome develops: the liver, spleen, and kidneys enlarge, and the accumulation of glucocerebroside in the cells of bone tissue and lungs destroys their structure.
What it is?
In short, Gaucher disease is a genetic disease in which fatty substances (lipids) accumulate in cells and on some organs. Gaucher disease is the most common of the lysosomal storage diseases. It is one of the forms of sphingolipidosis (a subgroup of lysosomal storage diseases), as it manifests itself in the dysfunction of sphingolipid metabolism.
The disorder is characterized by fatigue, anemia, low levels of platelets in the blood, and an enlarged liver and spleen. This is caused by a hereditary deficiency of the enzyme glucocerebrosidase, which acts on the fatty acid glucosylceramide. When the enzyme is damaged, glucosylceramide accumulates, in particular in white blood cells, most often in macrophages (mononuclear leukocytes). Glucosylceramide can accumulate in the spleen, liver, kidneys, lungs, brain and bone marrow.
Reasons for development
At the genetic level, mutations occur in the genes that are responsible for the production of the enzyme glucocerebrosidase. This gene with anomalies is localized on chromosome 1. These mutations cause low enzyme activity. Thus, glucocerebroside accumulates in macrophages.
Mesenchymal cells, called Gaucher cells, gradually grow and become hypertrophied. Since modifications occur in these cells, and they are located in the spleen, kidneys, liver, lungs, brain and bone marrow, they, in turn, deform these organs and disrupt their normal functioning.
Gaucher disease is an autosomal recessive disease. Therefore, any person can inherit a mutation of this enzyme with all the features in the same proportion, both from the father and from the mother. Thus, the degree of the disease and its severity will depend on the damage to the genes.
Theoretically, each person can inherit the glucocerebroside gene with lesions or completely healthy. As a result of inheriting a gene with abnormalities, a mutation of this enzyme occurs, but this does not yet indicate a disease. But when a child receives both affected genes, then a diagnosis of Gaucher disease is made. When inheriting one affected gene, a child is considered only a carrier of the disease, so there is a possibility of transmitting this trait, with hereditary pathology, to future generations. Thus, if both parents are carriers of the disease, a child can be born with Gaucher disease in 25% of cases, a carrier child in 50%, and a healthy child in 25%.
The frequency of occurrence of this hereditary pathology among ethnic races is 1:50,000, but it is much more often detected among Ashkenazi Jews.
Gaucher disease is also called a storage disease due to a deficiency of the enzyme, which should remove harmful metabolic products from the body, and not accumulate them. As a result of this, these substances accumulate in the macrophages of some organs and destroy them.

Classification and types of disease
The nature of the course of the disease varies in severity. Complications occur in childhood and adulthood. There are three types of disease:
- The first non-neuronopathic type. Sociology shows that it is common among Ashkenazi Jews. This pattern is called the Gaucher reaction. The clinical picture is characterized by a moderate, sometimes asymptomatic course of the disease. The psychology of behavior does not change, the brain and spinal cord are not damaged. Symptoms appear more often after thirty years of age. There are known cases of diagnosis in childhood. Timely treatment gives a favorable prognosis.
- The second type represents the neuronopathic infantile form and is rare. Symptoms appear in infancy as early as six months. Progressive damage to the child's brain occurs. Death can occur suddenly from suffocation. All children die before reaching two years of age.
- The third type (neuronopathic juvenile form). Symptoms have been observed since the age of 10 years. The intensification of symptoms is gradual. Hepatosplenomegaly - enlargement of the liver and spleen - is painless and does not impair liver function. Possible violation of behavioral psychology, the onset of neurological complications, portal hypertension, venous bleeding and death. Damage to bone tissue by Gaucher cells can lead to limited mobility and disability.
Symptoms of Gaucher disease
The clinical picture depends on the type of disease, but there are also general signs of this disease. You can suspect Gaucher syndrome (see photo) based on the following manifestations:
- pale skin;
- growth disturbance in children;
- general weakness;
- inflammation of the lymph nodes;
- enlarged liver and spleen;
- fractures in the absence of injuries;
- spontaneous nosebleeds;
- hemorrhagic stars on the skin.
Gaucher syndrome does not depend on the gender of the child. In addition, the symptoms of the disease often resemble the clinical picture of hematological pathologies. This makes diagnosing the disease difficult.
Characteristic signs of different forms of Gaucher syndrome:
Gaucher disease in children
Symptoms may begin to appear at different ages. The second type of disease often manifests itself at the age of six months. Patients in this case live up to 1-2 years. The third type is typical for children 2-4 years old, although it is sometimes observed in adolescence. The same applies to the first form. It can appear both in early childhood and adolescence. Symptoms of Gaucher syndrome in children:
- poor ability to suck and swallow;
- eye movement disorders;
- convulsions;
- breathing problems;
- whooping cough;
- yellow-brown skin pigmentation.
Diagnostics
Collecting anamnesis and complaints of the disease (clarification of the time of appearance of the first symptoms of the disease, how they progressed over time).
The disease can be suspected if an enlargement of the liver and spleen is accidentally detected (for example, according to ultrasound), suppression of the hematopoietic system (changes in blood tests: decreased levels of hemoglobin, platelets, the appearance of atypical blood cells), and the appearance of symptoms of bone damage.
At the next stage, special studies are carried out to confirm the diagnosis:
- Enzyme analysis - determination of the level of enzyme (glucocerebrosidase) in leukocytes and skin cells, which makes it possible to establish a diagnosis with absolute accuracy;
- biochemical blood test (decreased β-glucocerebrosidase activity, increased chitotriosidase levels);
- bone marrow examination (presence of characteristic Gaucher cells);
- molecular studies at the gene level (detection of genetic disorders);
- radiography and computer diagnostics (magnetic resonance imaging) of bones, since there may be areas of lower density that have specific signs for this disease.

Third type of Gaucher disease
How to treat Gaucher disease?
Specialized care for patients with the first and third types of the disease is aimed at eliminating symptoms and compensating for the primary genetic defect - increasing the amount of the missing enzyme, increasing the catabolism of glycosphingolipids. With type 2 pathology, therapeutic measures are not effective enough; doctors’ efforts are reduced to alleviating clinical manifestations - pain, cramps, respiratory disorders.
The general scheme includes the following areas:
- Enzyme replacement therapy. The main treatment method is lifelong enzyme replacement therapy (ERT) using recombinant glucocerebrosidase. The effectiveness is quite high - the symptoms are completely relieved, the quality of life of patients increases. ERT is appropriate for the third and first types of the disease. The drugs are administered intravenously. Frequent infusions sometimes cause inflammatory diseases of the veins (phlebitis).
- Substrate-reducing therapy. This direction is new in the treatment of Gaucher disease and is relatively widespread in the USA and European countries. Aimed at reducing the rate of production of the substrate glycosphingolipids and accelerating the catabolism of accumulating macromolecules. The drugs used are specific inhibitors of glucosylceramide synthase. The method is indicated for type 1 disease with mild to moderate symptoms.
- Symptomatic therapy. In cases of osteoporosis, complex therapy is prescribed, including taking calcium-containing medications, vitamin D and following a diet enriched with calcium. These measures can slow down bone loss, increase bone strength, and prevent fractures. For skeletal complications, analgesics (NSAIDs) and antibacterial therapy are used. Symptoms of neurological disorders can be relieved with antiepileptic drugs, nootropics, and muscle relaxants.
Prevention
The only method of preventing Gaucher disease is medical genetic counseling. If a family has a child suffering from this disease, the presence of glucocerebrosidase in the cells of the amniotic fluid is determined during subsequent pregnancies. If there is a deficiency of this enzyme in the fetus, doctors recommend termination of pregnancy.
Forecast
With the first type of disease, early diagnosis, and timely initiation of replacement therapy for Gaucher disease, positive dynamics are possible. The second type of glucocerebrosidosis is the most unfavorable, as it is more severe. Sick children usually do not live more than two years. The third form of Gaucher disease, with timely diagnosis and adequate treatment, allows maintaining the patient’s vital functions. Otherwise, he dies quite quickly from complications that develop.
Gaucher disease is a genetic disorder in which fatty substances (lipids) accumulate in cells and on some organs. Gaucher disease is the most common of the lysosomal storage diseases. It is one of the forms of sphingolipidosis (a subgroup of lysosomal storage diseases), as it manifests itself in the dysfunction of sphingolipid metabolism.
The disorder is characterized by fatigue, anemia, low levels of platelets in the blood, and an enlarged liver and spleen. This is caused by a hereditary deficiency of the enzyme glucocerebrosidase, which acts on the fatty acid glucosylceramide. When the enzyme is damaged, glucosylceramide accumulates, in particular in white blood cells, most often in macrophages (mononuclear leukocytes). Glucosylceramide can accumulate in the spleen, liver, kidneys, lungs, brain and bone marrow.
Manifestations of Gaucher disease may include an enlarged spleen and liver, severe neurological complications, swelling of the lymph nodes and adjacent joints, bloating, brownish skin color, anemia, low levels of platelets in the blood and sclera.
The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both men and women. Approximately 1 in 100 people in the world are carriers of Gaucher disease. The disease is named after the French physician Philippe Gaucher, who originally described it in 1882.
Types of Gaucher disease
Gaucher disease has three general clinical subtypes: type I, type II, and type III.
Type I is the most common form of the disease, occurring in 1 in 50,000 births. Symptoms of this type of Gaucher disease may appear early in life or in adulthood and include:
- Enlarged liver and significantly enlarged spleen;
- Weakness of skeletal bones;
- Anemia, thrombocytopenia and leukopenia;
- Kidney damage;
- Fatigue.
Type II usually begins to appear within the first six months of birth and occurs in approximately 1 in 100,000 births. Symptoms of this type of Gaucher disease include:
- Enlarged liver and spleen;
- Extensive and progressive brain damage;
- Eye movement disorders, spasticity, convulsions and limb rigidity;
- Weak ability to suck and swallow.
Affected children usually die by age 2 years.
Type III (chronic neuropathic form) can begin at any time, during childhood or even adulthood, and occurs in an incidence of 1 in 100,000 births. Main symptoms include an enlarged spleen or liver, seizures, incoordination, breathing problems, skeletal abnormalities, eye movement disorders, and blood disorders including anemia.
Symptoms of Gaucher disease
Common symptoms of Gaucher disease are:
- Painless hepatomegaly and splenomegaly - the size of the spleen can be from 1500 to 3000 ml, as opposed to the normal size of 50-200 ml. Splenomegaly can reduce appetite by putting pressure on the abdomen, and an enlarged spleen increases the risk of splenic rupture;
- Hypersplenism and pancytopenia – rapid and premature destruction of blood cells, leading to anemia, neutropenia, leukopenia and thrombocytopenia (with an increased risk of infection and bleeding);
- Cirrhosis of the liver;
- Severe pain in joints and bones, often in the hip and knee joints;
- Neurological symptoms;
- Type II: severe seizures, hypertension, mental retardation, apnea;
- Type III: muscle twitching, seizures, dementia, apraxia of the eye muscles;
- Osteoporosis;
- Yellowish-brown skin pigmentation.
Treatment of Gaucher disease
 Treatment of Gaucher disease subtypes 1 and 3 may begin with intravenous enzyme replacement with recombinant glucocerebrosidase, which can significantly reduce liver and spleen size, skeletal abnormalities, and reverse other manifestations. This procedure costs about $200,000 per patient and must be repeated annually for the patient's lifetime. Gaucher disease is also treated with the drug Velaglucerase Alfa, which has been approved as an alternative treatment since February 2010.
Treatment of Gaucher disease subtypes 1 and 3 may begin with intravenous enzyme replacement with recombinant glucocerebrosidase, which can significantly reduce liver and spleen size, skeletal abnormalities, and reverse other manifestations. This procedure costs about $200,000 per patient and must be repeated annually for the patient's lifetime. Gaucher disease is also treated with the drug Velaglucerase Alfa, which has been approved as an alternative treatment since February 2010.
Gaucher disease can also be treated with a successful bone marrow transplant, which treats non-neurological manifestations of the disease, since monocytes with active beta-glucosidase are injected during the procedure. However, this procedure carries significant risks and is rarely recommended for Gaucher disease.
Surgery to remove the spleen (splenectomy) is required in rare cases when the patient is anemic or when the enlarged organ is affecting the patient's health. Blood transfusion may occur for patients with symptoms of anemia. Also, in some cases, joint replacement surgery is required to improve mobility and quality of life.
Other treatments for Gaucher disease include antibiotics for infections, antiepileptics, bisphosphonates for bone lesions, and liver transplantation.
Gaucher disease is also treated with oral medications that act at the molecular level. Miglustat is one of these drugs and was approved for the treatment of Gaucher disease in 2003.
Video from YouTube on the topic of the article:
Today, Gaucher disease is one of the most common lysosomal diseases, in which there is an accumulation of lipids - fatty substances - in various cells and organs, which leads to their damage.
The disease has a genetic form of origin and significantly worsens the standard of living of patients.
Gaucher disease manifests itself in the form of dysfunction of the metabolic processes of sphingolipids responsible for the translation of cellular signals and cellular recognition. Nervous tissue is especially rich in sphingolipids, which explains its irreversible damage in this pathology.
As a result of a genetic mutation and inheritance of the affected gene, a deficiency of the enzyme glucocerebrosidase occurs, which promotes the decomposition of glucosylceramide of fatty acids. As a result, glucosylceramide accumulates in macrophages - mononuclear leukocytes and affects the site of accumulation. Such a storage reservoir can be the spleen, liver, bone and brain marrow, and lungs.
Gaucher's disease affects both men and women with equal frequency. For every hundred people in the population, at least one is a carrier of the pathological gene for the disease. The disease was first described by Philippe Gaucher, a French physician, at the end of the nineteenth century.
Types of inheritance of Gaucher disease
Observations and studies suggest two types of inheritance of the disease. The types of inheritance of Gaucher disease are divided according to their origin into autosomal dominant and autosomal recessive types.
With autosomal dominant inheritance, the child receives the pathology from one of the parents. Autosomal recessive inheritance occurs only when the affected gene is inherited from both parents.
In Gaucher disease, a predominance of the second type of inheritance is observed.
In addition, the patterns of inheritance of Gaucher disease are divided into three general subtypes, which have several distinctive features.
The first type of disease is the most common and occurs in 1 in 50,000 children born. Symptoms of Gaucher disease can appear both in childhood and in adulthood. These include kidney damage, enlarged spleen and liver, leukopenia, anemia and thrombocytopenia. Patients experience constant fatigue and weakness. There is weakening of skeletal bones.
Symptoms of type 2 Gaucher disease begin to appear in babies in the first half of the year - from 3 to 6 months. The development of the second type occurs half as often as the first. In addition to the enlargement of the spleen and liver, significant changes are observed in the brain, resulting in impaired motor functions of the eyes, convulsions and rigidity of the legs and arms, and decreased swallowing and sucking abilities. Unfortunately, children with this type of disease hardly survive to 2 years of age.
The chronic neuropathic form of the disease, defined as the third type, occurs with the same frequency as the previous one. Symptoms of Gaucher disease in this case are supplemented by respiratory failure, skeletal pathology, and blood diseases.
Common signs for all types of the disease may be a decrease in platelet levels, anemia, a brown tint to the skin, swelling of the lymph nodes and joints, and neurological abnormalities.
In Gaucher disease, an enlargement of the spleen to a significant size reduces appetite due to pressure on the abdomen. And further increase can lead to its rupture. The disease provokes destruction of the cellular composition of the blood, liver, mental retardation, and pain syndromes.
Diagnosis of Gaucher disease
 The disease is usually diagnosed without difficulty. But its symptoms can be mistaken for manifestations of similar diseases, so it is very important to make an accurate diagnosis.
The disease is usually diagnosed without difficulty. But its symptoms can be mistaken for manifestations of similar diseases, so it is very important to make an accurate diagnosis.
In this regard, the diagnosis of Gaucher disease should be carried out by exclusion.
To accurately determine the disease, enzyme activity is checked using a specialized blood test. The enzyme test is the most effective for identifying the disease.
The number of white blood cells, platelets and red blood cells is checked.
If necessary, DNA analysis is performed to determine the genetic background.
In addition to these forms of diagnosis of Gaucher disease, X-ray examinations, computed tomography, and nuclear magnetic resonance examination are performed to determine the general condition of the body. For neurological disorders, special testing is carried out using special tests.
Treatment of Gaucher disease
The most effective and expensive treatment for Gaucher disease types 1 and 3 is intravenous replacement of the enzyme with recombinant glucocerebrosidase. This procedure allows you to reduce the volume of the liver and spleen, reduce skeletal disorders and other anomalies of the body. As an alternative, use the drug Velaglucerase Alfa.
Bone marrow transplantation is used for non-neurological clinical diseases. But transplantation has a certain risk and is recommended only in exceptional cases.
Splenectomy - removal of the spleen, is also used only in cases of significant threat.
If there is progression, a blood transfusion is given. Sometimes surgical replacement of damaged joints or liver transplantation are performed.
Treatment of Gaucher disease with miglustat allows one to target the disease at the molecular level.
In addition, antibiotics, biosphosphonates and antiepileptic drugs are used in treatment.
RCHR (Republican Center for Health Development of the Ministry of Health of the Republic of Kazakhstan)
Version: Clinical protocols of the Ministry of Health of the Republic of Kazakhstan - 2016
Other sphingolipidoses (E75.2)
Orphan diseases
general information
Short description
Approved
Joint Commission on Healthcare Quality
Ministry of Health and Social Development of the Republic of Kazakhstan
from September 29, 2016
Protocol No. 11
Gaucher disease (GD)- lysosomal storage disease, a multisystem disease, it is based on a deficiency of the enzyme glucocerebrosidase, leading to a progressive increase in parenchymal organs, gradual infiltration of the bone marrow by macrophages loaded with lipids, profound disorders of hematopoiesis, and in a small part of patients damage to the central nervous system.
Correlation of ICD-10 and ICD-9 codes:
Date of development of the protocol: 2016
Usersprotocol: General practitioners, pediatricians, oncohematologists.
Level of evidence scale:
| A | A high-quality meta-analysis, systematic review of RCTs, or large RCTs with a very low probability (++) of bias, the results of which can be generalized to an appropriate population. |
| B | High-quality (++) systematic review of cohort or case-control studies, or High-quality (++) cohort or case-control studies with very low risk of bias, or RCTs with low (+) risk of bias, the results of which can be generalized to an appropriate population . |
| C | Cohort or case-control study or controlled trial without randomization with low risk of bias (+). The results of which can be generalized to the relevant population or RCTs with very low or low risk of bias (++ or +), the results of which cannot be directly generalized to the relevant population. |
| D | Case series or uncontrolled study or expert opinion. |
Classification
Classification
In accordance with the presence and characteristics of the clinical course and involvement of the central nervous system (CNS), they are divided into three types of Gaucher disease:
· Non-neuropathic (type I).
− Itype -bpainfulAndGaucher is the most common form of the disease in which the central nervous system is not affected (hence this type is also called non-neuropathic).
Symptoms are extremely varied - from asymptomatic forms to severe damage to organs and bones. In between these polar clinical groups there are patients with moderate enlargement of the spleen and almost normal blood composition, with or without bone lesions. Although this type of disease is sometimes called adult Gaucher disease, it can affect people of any age. The earlier clinical manifestations appear, the more severe the disease.
· Neuropathic (type II andIII).
− II type- acute neuronopathic. Type 2 Gaucher disease is a very rare, rapidly progressing disease characterized by severe damage to the brain, as well as almost all organs and systems.
Previously called Gaucher disease of newborns, type 2 disease is characterized by severe neurological disorders in the first year of a child’s life, including epileptic seizures, strabismus, muscle hypertonicity, and mental and physical development delays. Often this form of HD is combined with congenital ichthyosis. The disease occurs in less than 1 in 100,000 newborns. Progressive psychomotor degeneration ends in death, usually associated with respiratory failure.
− III type (chronic neuronopathic). Formerly called juvenile Gaucher disease, type 3 disease is characterized by slowly progressive damage to the brain as well as severe symptoms in other organs. This type of disease is also very rare. Signs and symptoms of type 3 Gaucher disease develop in early childhood and are similar to those of type 1 disease, except for signs of central nervous system involvement. Making an accurate diagnosis is only possible with the progression of neuropathy symptoms, confirmed by clinical studies. Patients with Gaucher disease type 3 who reach adulthood can live more than 30 years.
Diagnostics (outpatient clinic)
OUTPATIENT DIAGNOSTICS
Diagnostic criteria
Complaints and anamnesis:
weakness, increased fatigue;
· increased susceptibility to infections (respiratory infections, bacterial);
· manifestations of hemorrhagic syndrome (subcutaneous hematomas, bleeding of mucous membranes), and/or prolonged bleeding during minor surgical interventions;
· severe pain in bones and joints (nature and localization of pain, history of bone fractures);
· delayed physical and sexual development;
· manifestations of neurological symptoms (oculomotor apraxia or convergent strabismus, ataxia, loss of intelligence, sensory disturbances, etc.);
· family history (presence of splenectomy or the symptoms listed above in siblings, parents).
Increased abdominal volume
Physical examination:
· General inspection;
· Measurement of height, body weight, body temperature;
· Assessment of the condition of the osteoarticular system;
· Identification of signs of hemorrhagic syndrome;
· Detection of hepatosplenomegaly, lymphadenopathy;
· Assessment of the skin in the area of the knee and elbow joints (presence/absence of hyperpigmentation).
Clinical symptoms and signs of Gaucher disease depending on age
| System | Symptom | Newborns |
Children up to a year |
Children | Teenagers |
| CNS | Delay and regression of psychomotor skills | - | +++ | ++ | ± |
| convulsions | - | +++ | ++ | ± | |
| Skin | Collodion skin (swelling of the back of the feet and hands) | +++ | - | - | - |
| Gastrointestinal tract | Hepatosplenomegaly | ++ | +++ | +++ | +++ |
| Cirrhosis of the liver | - | - | - | - | |
| Ophthalmological | Abnormal eye movements | - | +++ | ++ | ± |
| Hematological | anemia | - | + | +++ | ++ |
| foam cells | ++ | +++ | +++ | +++ | |
| pancytopenia | - | + | + | + | |
| thrombocytopenia | - | + | +++ | +++ | |
| Skeletal | Bone pain | - | - | + | +++ |
| kyphosis | - | - | ± | ++ | |
| osteoporosis | - | - | ± | ++ | |
| Pathological fractures | - | - | ± | + | |
| Respiratory | Restrictive lung disease, pulmonary hypertension | - | ++ | ++ | + |
| Other | Early death | +++ | +++ | ± | - |
| Specific laboratory tests | β-D-glucosidase | ↓↓↓ | ↓↓ | ↓↓ | ↓↓ |
| Chitotriosidase |
Laboratory research :
· Detailed blood test: thrombocytopenia, leukopenia, anemia;
· BAK: increased levels of enzymes in the blood - ALT, AST, examination of iron metabolism (serum iron, TBSS, ferritin, transferrin) will help in the differential diagnosis between anemia of a chronic disease and an iron deficiency condition requiring standard treatment;
· Determination of the activity of the enzyme glucocerebrosidase and chitotriazidase in dried blood spots using tandem mass spectrometry or fluorimetry - to confirm the diagnosis;
· Molecular genetic research to confirm the diagnosis - identification of the glucocerebrosidase gene localized on the long arm of chromosome 1 (region 1q21q31);
· Morphological examination of the bone marrow helps to identify characteristic diagnostic elements - Gaucher cells and at the same time exclude the diagnosis of hemoblastosis or lymphoproliferative disease as the cause of cytopenia and hepatosplenomegaly.
Instrumental studies
Diagnostic algorithm

Algorithm for diagnosing Gaucher disease in children at the city and regional level

Algorithm for diagnosing Gaucher disease in children at the Republican level
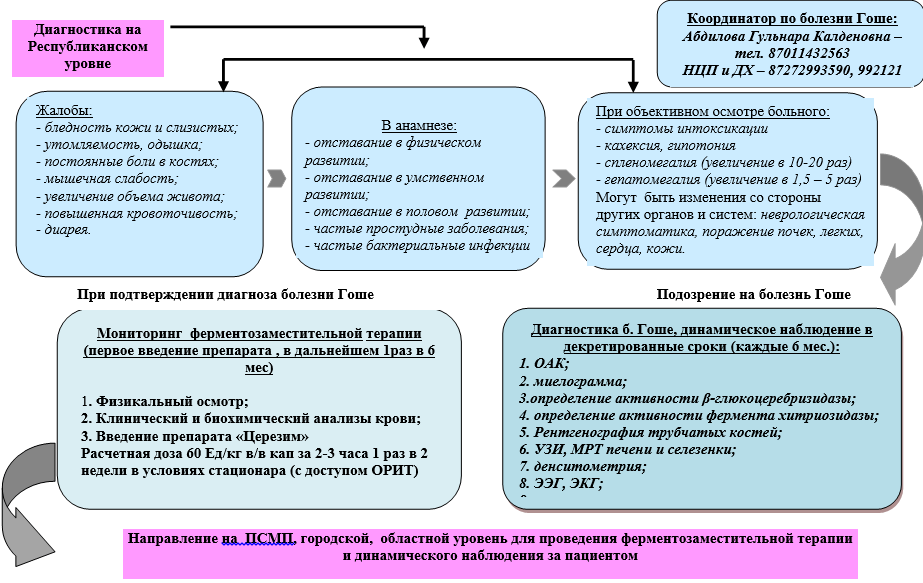
Diagnostics (hospital)
DIAGNOSTICS AT THE INPATIENT LEVEL
Diagnostic criteria:.
Diagnostic algorithm


List of basic diagnostic measures (UD - B)
detailed blood test
· blood chemistry
Determination of the activity of the enzyme glucocerebrosidase and chitotriazidase
· molecular genetic research
Ultrasound of the liver, spleen
MRI of the femur
· ECG
X-ray of skeletal bones
List of additional diagnostic measures:
· Myelogram - a bone marrow examination helps to identify characteristic diagnostic elements - Gaucher cells and at the same time exclude the diagnosis of hemoblastosis or lymphoproliferative disease as the cause of cytopenia and hepatosplenomegaly.
· CT scan of the lungs - to exclude pathology of the pulmonary system with prolonged neutropenia.
· MRI of the brain - for differential diagnosis with oncological diseases, excluding damage to the central nervous system in long-term cytopenic syndrome (risk of hemorrhagic stroke).
· MRI of the liver, spleen - in the presence of hepatosplenomegaly, there is a high risk of infarction of the liver and spleen due to the infiltration of organs and tissues with Gaucher cells.
· EchoCG - with severe tachycardia, against the background of symptoms of respiratory failure with prolonged cytopenic syndrome, there is a risk of complications from the cardiovascular system (exudative pericarditis, carditis, autonomic dysfunction).
· Coagulogram - in the presence of a cytopenic condition, the addition of a bacterial or viral infection, there is a possible risk of hemorrhagic condition, septic condition, disseminated intravascular coagulation syndrome.
· Dopplerography of the vessels of the portal system - to exclude portal hypertension.
Infectious complications against the background of long-term cytopenic syndrome areindication for additional laboratory tests:
· bacteriological examination of biological fluids,
· serological (virological) studies for CMV, Hepatitis B, C, (D), HIV, EBV,
· determination of C-reactive protein (quantitative),
· if transaminase levels increase: conduct serological (virological) studies to exclude viral hepatitis: CMV, A, B, C, EBV, if PCR results are positive
· coagulogram - study of hemostasis at the risk of septic complications, profuse hemorrhagic syndrome
· X-ray of skeletal bones - to identify and assess the severity of damage to the osteoarticular system (diffuse osteoporosis, characteristic flask-shaped deformation of the distal femur and proximal tibia (Erlenmeyer flask), foci of osteolysis, osteosclerosis and osteonecrosis, pathological fractures);
· Densitometry and magnetic resonance imaging (MRI) are more sensitive methods - they allow diagnosing bone damage (osteopenia, bone marrow infiltration) at early stages that are not visualized by radiography;
· Ultrasound and MRI of the liver and spleen make it possible to identify their focal lesions and determine the initial volume of organs, which is necessary for subsequent monitoring of the effectiveness of enzyme replacement therapy;
· Doppler echocardiography - in splenectomized patients;
esophagogastroduodenoscopy - in the presence of corresponding complaints or signs of portal hypertension.
Differential diagnosis
Differential diagnosis
Gaucher disease should be differentiated from all diseases that occur with hepatosplenomegaly, cytopenia, bleeding and bone damage.
| Diagnosis | Rationale for differential diagnosis | Surveys | Diagnosis exclusion criteria |
| Hemoblastoses and lymphomas | Hemorrhagic sm, bone pain, hepatosplenomegaly, |
2.myelogram, |
|
| Acquired aplastic anemia | Hemorrhagic sm, (+/_) bone pain, pancytopenia |
1. Complete blood count with platelet count, reticulocyte count, 2.myelogram, 3.molecular genetic blood test |
1. No decrease in the activity of the glucocerebrosidase enzyme and an increase in the activity of the chitotriazidase enzyme (in dried blood spots using tandem mass spectrometry or fluorimetry); 2. the glucocerebrosidase gene localized on the long arm of chromosome 1 (region 1q21q31) has not been identified; 3. Gaucher cells were not detected when counting cells in the myelogram |
| Chronic cholestatic liver diseases, liver cirrhosis as a result of chronic viral and non-viral hepatitis | Hepatosplenomegaly, increased levels of transaminases, bilirubin, cytopenic sm, hemorrhagic sm, painful sm |
1. Complete blood count with platelet count, reticulocyte count, 2.myelogram, 3.molecular genetic blood test 4. determination of the activity of the enzymes glucocerebrosidase and chytriosidase 5.B/x blood test 6. Ultrasound, CT, MRI of the abdominal organs |
1. No decrease in the activity of the glucocerebrosidase enzyme and an increase in the activity of the chitotriazidase enzyme (in dried blood spots using tandem mass spectrometry or fluorimetry); 2. the glucocerebrosidase gene localized on the long arm of chromosome 1 (region 1q21q31) has not been identified; |
| Chronic osteomyelitis, bone tuberculosis | Ossalgia, limitation of limb mobility |
2.myelogram, 3.molecular genetic blood test 4. determination of the activity of the enzymes glucocerebrosidase and chytriosidase 5.B/x blood test |
1. Absence of signs of cytopenia (decrease in hemoglobin, platelets, leukopenia), 2. No decrease in the activity of the glucocerebrosidase enzyme and an increase in the activity of the chitotriazidase enzyme (in dried blood spots using tandem mass spectrometry or fluorimetry); 3. the glucocerebrosidase gene localized on the long arm of chromosome 1 (region 1q21q31) has not been identified; 4.absence of hemorrhagic disease, 5. The characteristic club-shaped or flask-shaped swellings of the tibia (“Erlenmeyer flasks”) are not detected x-ray. 5.No hepatosplenomegaly |
| Other hereditary enzymopathies (Niemann-Pick disease |
Early onset of the disease (3-5 months), increase abdominal volume, delayed psychomotor development, convulsions, other neurological symptoms, abdominal pain, bleeding, emotional instability |
1.. Complete blood count with counting of platelets, reticulocytes, 2.myelogram, 3. molecular genetic study of blood (determination of mutations in the SMPD1, NPC1 and NPC2 genes, the glucocerebrosidase gene, localized on the long arm of chromosome 1 (region 1q21q31). 4. determination of the activity of the enzymes glucocerebrosidase and chytriosidase, sphingomyelinase 5.B/x blood test 6. Ultrasound, CT, MRI of the abdominal organs 7. X-ray examination of bone tissue (R, MRI, CT) 8.Examination by a neurologist |
1. No decrease in the activity of the glucocerebrosidase enzyme and an increase in the activity of the chitotriazidase enzyme (in dried blood spots using tandem mass spectrometry or fluorimetry); |
| Histiocytosis | Ossalgia, limited limb mobility, pancytopenia, hemorrhagic sm, hepatosplenomegaly, pneumonia, susceptibility to infections |
1. Complete blood count with platelet count, reticulocyte count, 2.myelogram, bone marrow immunophenotyping 3.molecular genetic blood test 4. determination of the activity of the enzymes glucocerebrosidase and chytriosidase 5. Blood test 6. Ultrasound, CT, MRI of the abdominal organs 7. X-ray examination of bone tissue (R, MRI, CT) |
1. No decrease in the activity of the glucocerebrosidase enzyme and an increase in the activity of the chitotriazidase enzyme (in dried blood spots using tandem mass spectrometry or fluorimetry); 2. the glucocerebrosidase gene localized on the long arm of chromosome 1 (region 1q21q31) is not detected; 3. The characteristic club-shaped or flask-shaped swellings of the tibia (“Erlenmeyer flasks”) are not detected x-ray. |
Treatment abroad
Get treatment in Korea, Israel, Germany, USA
Get advice on medical tourism
Treatment
Drugs (active ingredients) used in treatment
| Azithromycin |
| Alfacaltsidol |
| Amphotericin B |
| Acyclovir |
| Vancomycin |
| Voriconazole |
| Gentamicin |
| Diclofenac |
| Ibuprofen |
| Imiglucerase |
| Immunoglobulin G |
| Iodixanol |
| Caspofungin |
| Clindamycin |
| Kolekaltsiferol |
| Lactulose |
| Lornoxicam |
| Meropenem |
| Metronidazole |
| Micafungin |
| Ossein-hydroxyapatite complex |
| Paracetamol |
| Tramadol |
| Fluconazole |
| Cefotaxime |
| Ceftazidime |
| Ceftriaxone |
Treatment (outpatient clinic)
OUTPATIENT TREATMENT
Treatment tactics
Patients with all types (I, II, III) of Gaucher disease receive treatment on an outpatient basis.
Non-drug treatment:
· Regime - therapeutic and protective during the period of cytopenic syndrome, hemorrhagic, bone complications;
· Prevention of injuries, rehabilitation of chronic foci of infection;
· Psychological correction - psychotherapy, psychological adaptation.
Drug treatment
Modern treatment of GD consists of prescribing lifelong enzyme replacement therapy (ERT) with recombinant glucocerebrosidase, which relieves the main clinical manifestations of the disease, improving the quality of life of patients with GD and without causing significant side effects. . Every patient with clinical manifestations of GD (GD type 1, GD type 3) should be prescribed ERT. The dose of the drug should be selected individually in accordance with clinical and laboratory parameters. In connection with the development of laboratory diagnostics, when examining siblings (brothers and sisters of the proband), children with HD who do not have clinical manifestations can be identified. Such patients need to be monitored, but treatment should be started only when symptoms of the disease appear.
ERT is aimed at providing a sufficient amount of enzyme, which will allow the breakdown of deposits of unnecessary substances. Thus, enzyme replacement therapy works by supplementing or replacing the missing or defective enzyme in patients with Gaucher disease.

List of essential medicines
Imiglucerase
Pathogenetic treatment of Gaucher disease consists of lifelong administration of enzyme replacement therapy with recombinant glucocerebrosidase. The initial dose of imiglucerase per injection for type I GD is 30-40 units/kg without skeletal lesions and 60 units/kg in the presence of bone lesions. With type III in children, the dose can reach up to 100-120 units/kg .
The drug is administered intravenously at intervals of 1 time every 2 weeks. (2 times a month).
A stepwise dose reduction of 10-20 units/kg is possible with pronounced positive dynamics after 1 year of treatment for type 1 GD without bone damage and after 3-4 years with initial skeletal damage. Maintenance therapy: 15-60 units/kg intravenously, 3 hours every 2 weeks, for life.
Protocol for enzyme replacement therapy with Imiglucerase

List of additional medicines
· Paracetamol
Lornaxicam
Diclofenac
· Tramadol
Alfacalcidol
Flucanazole
Calcium Dz
Osteogenon
Acyclovir
Lactulose
· Cefotaxime
· Ceftazidime
· Ceftriaxone
Azithromycin
· Gentamicin
· Iodixanol
Meropenem
Nonsteroidal anti-inflammatory drugs:
· Paracetamol - tablets 200 mg, 500 mg; candles. Adults: 500 mg 3-4 times a day, for 3-7 days. Children at the rate of 60 mg/kg/day in 3-4 doses, 3-7 days;
· Ibuprofen tablets 200 mg, 400 mg; Children - ibuprofen 30-40 mg/kg/day,
· Lornaxicam - film-coated tablets 4 mg, 8 mg. Adults: 8 mg 2 times a day, orally, 2 weeks; lyophilisate for preparing a solution for intravenous and intramuscular administration, 8 mg. Adults: 8 mg 2 times a day, IM, 10 days;
· Diclofenac - solution for injection 2.5% in ampoules of 3 ml, tablets of 0.05 g, retard tablets of 0.025; 0.05 and 0.1 g; dragees, 0.025 g. Rectal suppositories, 0.05 and 0.1 g. Gel, cream, emulgel (1 g - 0.01 g ortofen) in tubes. Children: 2-3 mg/kg/day, IM, for 1-3-5 days. Adults: 7 mg 2 times a day, intramuscularly, 1-3-5 days.
· Tramadol - solution for injection 50 mg/ml, rectal suppositories 0.1 g, drops -2.5 mg/drop, capsules 50 mg. Orally, the usual initial dose for adults and children over 14 years of age is 50 mg (again, if there is no effect, after 30-60 minutes). Parenterally (IV, IM, SC) - 50-100 mg, rectally - 100 mg (re-introduction of suppositories is possible after 4-8 hours). The maximum daily dose is 400 mg (in exceptional cases it can be increased to 600 mg). Children aged 1 to 14 years orally (drops) or parenterally - a single dose of 1-2 mg/kg, maximum daily dose - 4-8 mg/kg.
Correctors of bone and cartilage tissue metabolism:
· Alfacalcidol, capsules 0.5 µg. The daily dose for adults varies from 0.07 mcg to 20 mcg, for children 0.01-0.08 mcg/kg, daily dose for children 0.01-0.08 mcg/kg.
· Calcium D3 - chewable tablets containing (active ingredients): calcium carbonate - 1250 mg (corresponds to 500 mg of elemental calcium); cholecalciferol - 200 IU (international units). Adults and children over 12 years of age - 2 tablets per day, preferably with meals.
· Osteogenon - tablets of ossein-hydroxyapatite complex - 830 mg; 2-4 tablets x 2 times a day.
Algorithm of action in emergency situations

Surgical intervention: No.
Other types of treatment:
· psychosocial rehabilitation: psychotherapy, psychological adaptation, environmental therapy;
· social adaptation and improvement of quality of life.
Indications for consultation with specialists :
| Specialist | Indication |
| traumatologist - orthopedist |
Excluding the presence of skeletal pathology in the child |
| Neuropathologist, psycho-neurologist | assessment of neurological status, neuropsychic status, determination of the type of disease |
| physiotherapist |
determination of physiotherapeutic treatment methods |
| physical therapy doctor | selection of an individual physical therapy program |
| geneticist | diagnosis confirmation, genotyping |
| If necessary, consultation with other specialists is possible depending on the clinical case. | |
Preventive actions:
· early diagnosis of clinical manifestations of Gaucher disease to prevent complications;
· medical genetic counseling to clarify genetic risk.
· prevention of infectious complications against the background of long-term cytopenic syndrome, which in some cases are the main cause and in some cases even the death of the patient.
· oral care: 6-10 times a day, rinsing the mouth with disinfectant solutions intended for treating the oral mucosa. Thorough but gentle care for teeth and gums; limiting the use of even soft toothbrushes; give preference to oral shower; in case of thrombocytopenia or vulnerable mucous membranes, the use of toothbrushes should be excluded; instead, additional treatment of the mouth with astringents is necessary.
If signs of stomatitis appear, the following must be added to basic therapy:
· Fluconazole - estimated dose 4-5 mg/kg per day, capsules 50 mg, 100 mg, 150 mg, solutions for infusion 2 mg/ml, gel for treating the mouth r.o.
· Acyclovir - calculated dose 250 mg/m 2 x 3 times a day, tablets 200 mg, injection solution 250 mg, ointment for external use.
· If defects appear in the oral mucosa: avoid using toothbrushes
2) with the development of widespread necrotizing stomatitis, systemic antifungal and antibacterial therapy is indicated:
· Cefotaxime, 1 g bottle for solution preparation. Adults 1-2g, 2-3 times a day, intravenously, intramuscularly, 7-10 days. Children 50-100 mg/kg body weight/day, 2-4 times a day, IM, IV, 7-10 days;
· Ceftazidime, bottle 250 mg, 500 mg, 1 g, 2 g for solution preparation. Adults: 1-6 g/day in 2 or 3 doses IV or IM. Children over 2 months: 30-100 mg/kg/day in 2-3 divided doses, with reduced immunity - up to 150 mg/kg/day (6 g/day maximum) in 3 divided doses. Newborns and infants up to 2 months: 25-60 mg/kg/day in 2 divided doses.
· Ceftriaxone, 500 mg bottle, 1 g for solution preparation. Children 50-80 mg/kg/day IV drops 1 hour for 7-10 days;
· Iodixanol, solution for injection, 100 mg/2 ml and 500 mg/2 ml. Adults and children over 12 years of age are prescribed IM, IV (stream, over 2 minutes or drip) 5 mg/kg every 8 hours or 7.5 mg/kg every 12 hours for 7-10 days.
· Gentamicin, solution for injection, ampoules 40 mg/ml. adults 3-5 mg/kg (maximum daily dose) in 3-4 doses, 7-10 days. It is prescribed to young children only for health reasons in case of severe infections. The maximum daily dose for children of all ages is 5 mg/kg.
· Azithromycin, capsules 250, 500 mg. For children weighing more than 10 kg at the rate of: on day 1 - 10 mg/kg body weight; in the next 4 days - 5 mg/kg. A 3-day course of treatment is possible; in this case, the single dose is 10 mg/kg. (Course dose 30 mg/kg body weight). Adults with infections of the upper and lower respiratory tract, infections of the skin and soft tissues are prescribed 0.5 g on the 1st day, then 0.25 g from the 2nd to the 5th day or 0.5 g daily on for 3 days (course dose 1.5 g).
· Meropenem, powder for the preparation of a solution for intravenous administration, 0.5 and 1.0 g. For children aged 3 months to 12 years, the recommended dose is 10-20 mg/kg every 8 hours, depending on the type and severity of infection, the sensitivity of the pathogen and the patient's condition. In children weighing more than 50 kg, the adult dose should be used.
3) Intestinal decontamination is carried out at the choice of the hospital; decontamination may be refused. Decontamination (preventive therapy) is recommended for initial intestinal lesions. For selective intestinal decontamination:
Ciprofloxacin at a dose of 20 mg/kg per day, 100 mg in a bottle, 250 mg, 500 mg in tablets, eye drops, ear drops;
4) It is mandatory for everyone caring for the patient - parents and visitors - to maintain personal hygiene, and constantly wash their hands.
Substitution therapy tactics and according to Order No. 666 “On approval of the Nomenclature, Rules for the procurement, processing, storage, sale of blood, as well as Rules for the storage, transfusion of blood, its components and blood products dated March 6, 2011, Appendix to Order No. 417 Order dated May 29. 2015.
Patient monitoring:
· lifelong ERT;
dynamic control: 1 year - once every 3 months, then once every 6 months:
· social adaptation;
· observation by a geneticist of the family of a HD patient.
Indicators of treatment effectiveness:
· improvement/stabilization of hematological parameters (relief of cytopenic syndrome, lack of dependence on blood transfusions);
· restoration of glucocerebrosidase levels, reduction of chitotriosidase levels;
· elimination of pain;
· restoration of bone tissue;
· improvement/stabilization of the function of extra-abdominal organs (heart, lungs, eyes);
· reducing the frequency of respiratory infections;
· reducing the rate of disease progression;
improving the patient’s quality of life (restoration of mental, spiritual, physical development).
Treatment (ambulance)
DIAGNOSTIC MEASURES CARRIED OUT AT THE EMERGENCY CARE STAGE
Diagnostic measures:
· taking anamnesis;
· physical examination;
· determination of cardiac pathology (pulse oximetry, blood pressure, heart rate, ECG).
Drug treatment
Cardiopulmonary resuscitation as indicated;
· syndromic-symptomatic therapy according to indications;
· oxygen therapy;
· prevention of aspiration;
· analgesic anti-inflammatory therapy
Treatment (inpatient)
INPATIENT TREATMENT
Treatment tactics:see outpatient level.
Drug treatment: see outpatient level.
Drug treatment is carried out in accordance with the Clinical Protocols for serious complications.
Drug therapy is intensified when complications arise against the background of long-lasting cytopenic syndrome, layering of a viral/bacterial infection, or progression of the underlying disease. The most serious life-threatening complications are infectious complications. The presence of fever in a patient with neutropenia (neutrophils< 500/мкл) считается однократное повышение температуры тела >37.9 0 With a duration of more than an hour or several rises (3 - 4 times a day) up to 38 0 C. Taking into account the high risk of fatal infection, fever in a patient with neutropenia is regarded as the presence of infection, which dictates the immediate start of empirical antibacterial therapy and conducting an examination to clarify the nature of the infection. Many initial antibacterial regimens have been proposed, the effectiveness of which is generally identical.
General provisions:
· when choosing a starting combination of antibiotics, it is necessary to take into account: the results of repeated bacteriological studies in this clinic in other patients; duration of current neutropenia, infectious history of the patient, previous courses of antibiotics and their effectiveness
· along with the appearance of fever, all other clinical data: arterial hypotension, unstable hemodynamics are an indication for the immediate prescription of a combination of antibiotics: carbopenems (meropenem (or imipenem/cilastatin)) + aminoglycoside (amikacin) + vancomycin.
· long standing CVC and fever after its rinsing and/or not just fever, but amazing chills ®Vancomycin is already in the starting combination;
· clinic of enterocolitis with diarrhea: to the initial combination - vancomycin per os 20 mg/kg per day. It is possible to prescribe Metronidazole (per os and/or i.v.)
· severe stomatitis with inflammatory changes in the gums ® penicillin, clindamycin in combination with beta-lactam or Meropenem/
characteristic rash and/or the presence of fungal drusen in the urine and/or characteristic lesions in the liver and spleen during sonography®
· Amphotericin B - lyophilisate for solution preparation. The starting dose is 0.5 mg/kg on the 1st day, the next day - the full therapeutic dose is 1 mg/kg per day once. When using Amphotericin B, it is necessary to monitor kidney function and do a biochemical blood test (electrolytes, creatinine). Constant correction of potassium to normal values is necessary. During the infusion of Amphotericin B, as well as for approximately 3-4 hours after the infusion, reactions to the administration of the drug in the form of fever, chills, and tachycardia, which can be relieved with analgesics, may be observed. If renal function is impaired, it is necessary to use voriconazole, Cancidas, and lipid forms of amphotorecin B.
· Voriconazole - 50 mg tablet, lyophilisate for solution 200 mg/bottle. SD 4-6 mg/kg.
Caspofungin - lyophilisate for the preparation of solution for infusion 50 mg
Micofungin - lyophilisate for the preparation of solution for infusion 50 mg
Change of antibiotics taking into account the sensitivity of the isolated flora. The effectiveness of initial antibiotic therapy should be assessed after 72 hours; however, a detailed examination of such a patient at intervals of 8-12 hours is always necessary, assessing hemodynamic stability and the degree of intoxication, and the appearance of new infectious foci. Antibacterial therapy continues until neutropenia resolves and all infectious foci are completely resolved.
In case of deep aplasia, the risk of developing septic complications, passive immunization with Immunoglobulins G - 0.1-0.2 g/kg/day IV drops.
List of essential medicines:
Imiglucerase 30-60 units/kg IV drop for 3 hours
List of additional medicines:
· Paracetamol
Lornaxicam
Diclofenac
· Tramadol
Alfacalcidol
Flucanazole
Calcium Dz
Osteogenon
Acyclovir
Lactulose
· Cefotaxime
· Ceftazidime
· Ceftriaxone
Azithromycin
· Gentamicin
· Iodixanol
Meropenem
Immunoglobulin G
Amphotericin B
Voriconazole
Caspofungin
Micofungin
Vancomycin
Metronidazole
· Clindamycin
Surgical intervention:
· correction of pathological fractures of bone tissue, contractures in the joint.
Other types of treatment:
· physical rehabilitation: physiotherapy, therapeutic exercises, massage;
· psychosocial rehabilitation: psychotherapy, psychological adaptation, environmental therapy.
Indications for consultation with specialists: see outpatient level.
Indications for transfer to the intensive care unit:
· decompensated condition of the patient;
· generalization of the process with the development of complications requiring intensive monitoring and therapy;
· postoperative period;
· development of complications during intensive chemotherapy, requiring intensive treatment and observation.
Indicators of treatment effectiveness:
· restoration of mental, spiritual, physical development;
· restoration of mobility and performance;
· elimination of pain during the first 2 years of therapy;
· prevention of bone crises;
· prevention of the development of osteonecrosis and subchondral collapse;
· improvement of bone mineral density;
· increase in bone mineral density over 3 years of therapy;
· achieving normal growth rates according to population standards within 3 years of therapy;
· reaching the normal age of puberty.
· normalization of blood counts during the first 3 years of therapy;
· reduction of hepatosplenomegaly;
· improvement of the condition of extra-abdominal organs (heart, lungs, eyes).
Further management:
When the condition is stabilized, hematological parameters are restored, pain, intoxication, and hemorrhagic symptoms are relieved, the child is discharged for outpatient treatment under the supervision of a pediatrician or local hematologist to continue enzyme replacement therapy under the supervision of tests. Further monitoring of the patient's condition is carried out at the outpatient level.
Hospitalization
Indications for planned hospitalization
Planned hospitalization in a hospital is indicated to verify the diagnosis and to adjust the dose of enzyme replacement therapy.
Indications for emergency hospitalization
· Cytopenic syndrome;
· Severe pain syndrome (“bone crisis”);
· Pathological fracture of skeletal bones;
· Respiratory failure.
Information
Sources and literature
- Minutes of meetings of the Joint Commission on the Quality of Medical Services of the Ministry of Health of the Republic of Kazakhstan, 2016
- 1) Zub N.V. “Gaucher’s disease: prevalence, semiotics, quality of life and clinical and economic rationale for enzyme replacement therapy” abstract by Ph.D. Moscow 2010 2) Lukina E.A. “Gaucher's disease: current state of the issue” Russian Medical News 2008, Volume XIII, No. 2 p. 51-56. 3) Belogurova M.B. "Pathogenesis, clinical picture, diagnosis and treatment of Gaucher disease." Pediatrics and pediatric surgery. No. 3 2010, pp. 43-48. 4) Aerts J.M., van Weely S., Broot R., et al. Pathogenesis of lysosomal storage disorders as illustrated by Gaucher disease // J. Inher. Metab. Dis. – 1993. – Vol. 16. No. 2. – P.288-291. 5) Beutler E., Grabowski G.A., Scriver C.R., et al. The metabolic and molecular bases of inherited disease //McGraw-Hill, New York, 2001. – P.3635-3668. 6) de Frost M., vom Dahl S., Weverling G.J., et al. Increased incidence of cancer in adults Gaucher disease in Western Europe // Blood Cells Mol. Dis. – 2006. – Vol. 36.– P.53-58. 7) Taddei T.H., Kacena K.A., Yang M., et al. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients // Am. J. Hematol. – 2009. – Vol. 84. No. 4. – P.208-214. 8) Niederau C. Gaucher disease. Bremen:UNI-MED; 2006. 84 p. 9) Zimran A., Kay A., Beutler E. et al. Gaucher disease: clinical, laboratory, radiologic and genetic features of 53 patients. Medicine 1992; 71: 337–53. 10) Weinreb N. J. Type I Gaucher disease in elderly patients. Gaucher Clin. Persp. 1999; 7(2): 1–8. 11) Vorobyov A.I. (ed.) Rational pharmacotherapy of diseases of the blood system. M.: Littera, 2009, 563–6. 12) A.V. Davydova “Lysosomal storage diseases: Gaucher disease” Siberian Medical Journal, 2009, No. 5. P.9-14. 13) Mikosch P., Reed M., Baker R., et al. Changes of bone metabolism in seven patients with Gaucher disease treated consecutively with imiglucerase and miglustat // Calcif. Tissue Int. – 2008. – Vol. 83, No. 1. – P.43-54. 14) vom Dahl S., Poll L., Di Rocco M., et al. Evidencebased recommendations for monitoring bone disease and the response to enzyme replacement therapy in Gaucher patients // Current Med. Research and Opinion. – 2006. –Vol. 22. No. 6. – P.1045-1064. 15) Wenstrup R.J., Roca-Espiau M., Weinreb N.J., et al. Skeletal aspects of Gaucher disease: a review // Br. J. Radiol. – 2002. – Vol. 75. – P.2-12. 16) Cox TM, Schofield JP. Gaucher's disease: clinical features and natural history. Bailliere's Clinical Haematology. 1997;10(4): 657-689. 17) Grabowski G. Gaucher disease: Enzymology, genetics, and treatment. In:Harris H, Hirshchorn K, eds. Advances in Human Genetics. New York, NY: Plenum Press; 1993;21: 377-441. 18) Vorobyov A.I. (ed.) Rational pharmacotherapy of diseases of the blood system. M.: Littera, 2009, 563–6. 19) NIH Technology Assessment Panel on Gaucher Disease. Gaucher disease: current issues in diagnosis and treatment. JAMA. 1996;275:548-553. NIH Technology Assessment Panel on Gaucher Disease. Gaucher disease: current issues in diagnosis and treatment. JAMA. 1996;275:548-553. 20) Grabowski G. A. Phenotype, diagnosis, and treatment of Gausher’s disease // Lancet.-2008.- Vol. 372.No.9645.-R. 1263-1271. 21) Abdilova G.K., Boranbaeva R.Z., Omarova K.O. et al. “Modern diagnosis and treatment of Gaucher disease in children in Kazakhstan” methodological recommendations, Almaty 2015 pp. 26-27. 22). Physician’s guide to the diagnosis, treatment and follow-up of inherited metabolic diseases, ed. N.Blau, M.Duran, K.M. Gibson, C. Dionisi-Vici. 2014) 23) “Federal clinical guidelines for the provision of medical care to children with Gaucher disease” Moscow, 2015
Information
ABBREVIATIONS USED IN THE PROTOCOL:
ALT - alanine aminotransferase
AST - aspartic aotocolaminotransferase
GD - Gaucher disease
MRI - magnetic resonance imaging
CBC - complete blood count
OAM - general urine analysis
Ultrasound - ultrasound examination
ERT - enzyme replacement therapy
ECG - electrocardiogram
EchoCG - echocardiography
LSD - lysosomal storage diseases
CNS - central nervous system
DNA - deoxyribonucleic acid
HS - hemorrhagic syndrome
ESR - erythrocyte sedimentation rate
CT-computed tomography
LIST OF PROTOCOL DEVELOPERS:
1) Boranbaeva Riza Zulkarnaevna - Doctor of Medical Sciences, Director of the Republican State Enterprise "Scientific Center of Pediatrics and Children's Surgery".
2) Gulnara Kaldenovna Abdilova - Candidate of Medical Sciences, Deputy Director of the Republican State Enterprise "Scientific Center of Pediatrics and Pediatric Surgery" in pediatrics.
3) Omarova Kulyan Omarovna - Doctor of Medical Sciences, Professor, Chief Researcher of the Republican State Enterprise "Scientific Center of Pediatrics and Children's Surgery".
4) Manzhuova Lyazat Nurbapaevna - Candidate of Medical Sciences, Head of the Department of Oncohematology for Older Children of the Republican State Enterprise "Scientific Center of Pediatrics and Pediatric Surgery".
5) Elmira Maratovna Satbaeva - Candidate of Medical Sciences, RSE at the PME "Kazakh National Medical University named after S.D. Asfendiyarov", head of the Department of Pharmacology.
NOTICE OF NO CONFLICTS OF INTEREST: are missing.
REVIEWERS:
1. Kurmanbekova Saule Kaspakovna - Professor of the Department of Internship and Residency in Pediatrics No. 2 of the Kazakh National Medical University. S.D. Asfendiyarova.
INDICATING THE CONDITIONS FOR REVISING THE PROTOCOL: revision of the protocol 3 years after its entry into force and/or when new diagnostic/treatment methods with a higher level of evidence become available.
Attached files
Attention!
- By self-medicating, you can cause irreparable harm to your health.
- The information posted on the MedElement website and in the mobile applications "MedElement", "Lekar Pro", "Dariger Pro", "Diseases: Therapist's Guide" cannot and should not replace a face-to-face consultation with a doctor. Be sure to contact a medical facility if you have any illnesses or symptoms that concern you.
- The choice of medications and their dosage must be discussed with a specialist. Only a doctor can prescribe the right medicine and its dosage, taking into account the disease and condition of the patient’s body.
- The MedElement website and mobile applications "MedElement", "Lekar Pro", "Dariger Pro", "Diseases: Therapist's Directory" are exclusively information and reference resources. The information posted on this site should not be used to unauthorizedly change doctor's orders.
- The editors of MedElement are not responsible for any personal injury or property damage resulting from the use of this site.
