Болест на Гошеобичайно е да се нарича нарушение на метаболизма на сфинголипидите, което е отговор на дефицит на ензим, който разрушава глюкоцеребразид, такова усложнение може да доведе до отлагане на глюкоцереброзид. Най -честите симптоми на болестта на Гоше са хепатоспленомегалия или промени в централната нервна система. За да се диагностицира правилно заболяването, е необходимо да се проведат цитохимични изследвания на левкоцитите.
Това е заболяване, което не се среща толкова често, предава се наследствено, когато и двамата родители са носители на дефектния ген. За първи път болестта на Гоше видя бял свят на страниците на медицинските ръководства през 1882 г.
Липсата на ензим бета-глюкоцереброзидаза в заобиколени от мембрани клетъчни органели може да доведе до образуването на голямо количество хранителна среда за микроорганизми от тази органична материя в клетките на макрофаговата система на целия организъм, като правило този процес възниква и се развива в жлезите, както и в клетките на костния мозък и далака.
Към днешна дата науката е установила три типа болест на Гоше:
- Тип 1 се среща най -често при хора, преминали етапа на пубертета, и също е постоянен, този тип не може да се характеризира с наличието на невронопатия. Болест тип 1 може да се нарече най -бавният и често срещан тип, при който централната нервна система няма да бъде засегната.
- Тип 2, при който децата стават мишени на увреждане, не е толкова разпространен в науката. При този вид заболяване, като правило, се засягат неврони, което води до почти пълна атрофия на цялата нервна система. С тази диагноза детето умира още като бебе.
- Тип 3 в науката обикновено се нарича "ювенилен", като при този тип симптомите на процеса са по -слабо изразени, в този случай атрофията на невронните клетки е неизбежна. Трябва да се отбележи, че вид 3 също е доста рядък. Учените характеризират този вид заболяване чрез постепенното, както и хаотичното привързване на цялата нервна система към този процес.
Фактът, че болестта на Гоше може да съществува в различни външни форми, както и в условия, при които се наблюдават различни вътрешни структури, потвърждава разнообразието от промени в силно структурирания ген на глюкоцереброзидаза на хромозома 1. Въпреки това могат да бъдат проследени заболявания с различна тежест сред един даден генотип ... Основното място във въпроса за силата на трансформация е отредено на рязко увеличаване на броя на макрофагите в органи и тъкани, което е отговор на появата на голямо количество глюкоцереброзид, но методите на неговото функциониране не са известен.
Зиготата на Гоше, като правило, е подобна на овална и има размер около 70-80 мм в диаметър, както и бледа цитоплазма. Той съдържа две или повече ядра с повишена пигментация, които са изместени към периферията. В средата на тези ядра са нишковидни протеинови структури, които са разположени едновременно една спрямо друга.
В процеса на развитие на болестта се натрупва бета-глюкоцереброзид, който в крайна сметка получава своя произход от разпаднали се плазмалеми, има тенденция да се превърне в утайка в мембранните клетъчни органели и да образува удължени подобия на тръби с размер двадесет, а понякога и с дължина четиридесет мм, тези тръби могат да се видят с увеличение от 2-3 хиляди пъти. Такива зиготи могат да бъдат открити в CML, както и в тумор на В-лимфоцитната система, тъй като в резултат на тези заболявания се наблюдава ускорен процес на обмен на бета-глюкоцереброзид.
Симптоми на болестта на Гоше
При нормални условия се наблюдава органично вещество, което разрушава глюкоцеребразид, който хидролизира глюкоцереброзиди, като същевременно образува глюкоза и керамиди. Ако по време на развитието на организма е имало увреждане на органичната материя, получено на генетично ниво, това може да доведе до факта, че клетките започват да улавят и усвояват твърди частици, като по този начин създават зиготи на Гоше. Натрупване на тези зиготи в пространства
около съдовете в субстанцията на човешкия мозък провокира процеса на заместване на мъртви или заменени неврони с глиални клетки. Има 3 вида, които се различават по модела на възникване и разпространение на заболявания с различна етиология, активността на органичните вещества, а също и по естеството на проявите:
Тип 1 се характеризира с най -висока честота на поява - този тип се среща при 90% от популацията (не е невронопатичен).
Активността, която може да се нарече остатъчна, наблюдавана в органичните вещества, има най -висок процент. Първите прояви могат да се появят в периода от 2 години до старост. Основните симптоми са промени в костните клетки, забавено развитие по отношение на физиологията, забавена активност по време на пубертета, кръвоизлив в кожата. Последният симптом, придружен от назални кръвоизливи, е доста често срещан. След като направят рентгенова снимка, като правило лекарите установяват, че окончанията на дългите кости са разширени, а костната плоча на устната кухина е изтънена.
Тип 2 се характеризира с най -ниската честота на възникване (остра невронопатична). При този тип се наблюдава намаляване на остатъчната активност на органичните вещества. Първите сериозни признаци могат да бъдат открити в ранна възраст - след раждането. Основните симптоми са бързо развиващите се неврологични разстройства: нееластичността, за съжаление, този тип в повечето случаи води до смърт на възраст около две години.
Тип 3 е между най -често срещаните и най -редките (подостър невронопатичен). Жизнената активност на органичните вещества, както и тежестта на заболяването, съответно, са междинни между типове 1 и 2. Първите симптоми на този тип могат да бъдат открити в детството. Клиничните прояви могат да променят показателите си в зависимост от сорта, а също така включват, както и нарушена координация на движенията (Ilia), инфекция на органи и костни тъкани (Nib) и дегенеративни заболявания на централната нервна система с непрозрачност на роговицата (III) . Ако при този тип пациентът преживее юношеския стадий, в бъдеще той, като правило, живее дълго време.

Диагностика на болестта на Гоше
Диагнозата на това заболяване обикновено се състои в цитохимичното изследване на левкоцитите. Видовете, както и носителят, обикновено се идентифицират въз основа на анализа на естеството на мутациите. Зиготите на Гоше имат диагностична стойност.
ВИДЕО
Лечение на болестта на Гоше
За типове 1 и 3 се препоръчва заместващо лечение със специални комплексни лекарства, използващи плацентарна или рекомбинантна глюкоцереброзидаза; с лечение тип 2, за съжаление, е безполезно, освен това е напълно непознато за науката и медицината. По време на лечението настъпва промяна в ензима, която се извършва за неговия бърз и навременен транспорт до клетъчния органоид, заобиколен от мембрана. На пациентите, лекувани със специални комплексни лекарства, се предписва ежедневно проследяване на нивото на багрилото в кръвта, както и безцветни кръвни клетки; постоянно наблюдение на размера на черния дроб и далака с помощта на CT или MRI; непрекъснато наблюдение на костни лезии с пълно наблюдение на цялата скелетна система, рентгеново абсорбциометрично сканиране с дуал-енергия или ЯМР.
По правило на пациентите се предписват следните лекарства: Miglustat, който трябва да се приема в определени дози, а именно три пъти дневно, сто mg перорално, miglu -stat - този вид лекарства намалява концентрацията на глюкоцереброзид, а също се превръща в своеобразно решение за пациенти, които според някои поради причини, поради които не могат да се подложат на лечение с ензимна заместителна терапия.
Обикновено се предписва на пациенти с анемия, както и с намаляване на броя на левкоцитите и тромбоцитите в кръвта, както и в случая, когато далакът нараства по размер, което започва да причинява дискомфорт.
За цялостно лечение на пациенти с това заболяване лекарите прибягват до стволови клетки или стволови клетки, но този вид лечение е най -опасен за здравето и живота на пациента, поради което се използва възможно най -рядко.
Заболяването принадлежи към лизозомни заболявания на съхранение (глюкозилцерамидна липидоза).
Характеризира се с дефицит на ензима глюкоцереброзидаза. Това води до метаболитни нарушения. Липидите не се разграждат до продукти за повторна консумация; глюкоцереброзид се натрупва в клетките на макрофагите. Те се увеличават, придобиват характерния вид сапунени мехурчета и се утаяват в тъканите на тялото.
Развива се синдром на Гоше: черният дроб, далакът, бъбреците се увеличават и натрупването на глюкоцереброзид в клетките на костните тъкани и белите дробове разрушава тяхната структура.
Какво е?
Накратко, болестта на Гоше е генетично заболяване, при което мастните вещества (липиди) се натрупват в клетките и върху някои органи. Болестта на Гоше е най -често срещаното заболяване на лизозомното съхранение. Това е една от формите на сфинголипидоза (подгрупа от заболявания на лизозомното съхранение), тъй като се проявява в дисфункционалния метаболизъм на сфинголипидите.
Разстройството се характеризира с умора, анемия, ниски кръвни тромбоцити и увеличен черен дроб и далак. Това се дължи на наследствен дефицит на ензима глюкоцереброзидаза, който действа върху глюкозилцерамид на мастни киселини. Когато ензимът е повреден, глюкозилцерамид се натрупва, по -специално в левкоцити, най -често в макрофаги (моноядрени левкоцити). Глюкозилцерамидът може да се натрупва в далака, черния дроб, бъбреците, белите дробове, мозъка и костния мозък.
Причини за развитие
На генетично ниво се появяват мутации в гени, които са отговорни за производството на ензима глюкоцереброзидаза. Този ген с аномалии е локализиран на 1 -ва хромозома. Тези мутации причиняват ниска ензимна активност. По този начин има натрупване на глюкоцереброзиди в макрофагите.
Мезенхимните клетки, наречени клетки на Гоше, постепенно растат и стават хипертрофирани. Тъй като в тези клетки настъпват промени и те се намират в далака, бъбреците, черния дроб, белите дробове, мозъка и костния мозък, те от своя страна деформират тези органи и нарушават нормалното им функциониране.
Болестта на Гоше е автозомно рецесивно разстройство. Следователно всеки човек може да наследи мутация на този ензим с всички характеристики в същото съотношение, както от бащата, така и от майката. По този начин степента на заболяването и неговата тежест ще зависят от увреждането на гените.
На теория всеки може да наследи глюкоцереброзидния ген с лезии или напълно здрав. В резултат на наследяването на ген с аномалии настъпва мутация на този ензим, но това не означава заболяване. Но когато едно дете получи и двата засегнати гена, тогава се диагностицира болестта на Гоше. С наследяването на един засегнат ген детето се счита само за носител на болестта, поради което съществува възможност за предаване на тази черта с наследствена патология на бъдещите поколения. Така при двамата родители, носещи болестта, едно дете може да се роди с болест на Гоше в 25% от случаите, дете носител - в 50% и здраво - в 25%.
Честотата на появата на тази наследствена патология сред етническите раси е 1: 50 000, но е много по -често срещана сред ашкенази евреите.
Болестта на Гоше се нарича още болест на натрупване поради липсата на ензим, който трябва да премахва вредните метаболитни продукти от тялото, а не да ги натрупва. В резултат на това тези вещества се събират в макрофагите на някои органи и ги унищожават.

Класификация и видове заболявания
Характерът на хода на заболяването е с различна тежест. Усложненията възникват в детска и зряла възраст. Има три вида заболяване:
- Първи невронопатичен тип. Социологията показва, че е често срещана сред евреите ашкенази. Този модел се нарича реакция на Гоше. Клиничната картина се характеризира с умерено, понякога безсимптомно протичане на заболяването. Психологията на поведението не се променя, мозъкът и гръбначният мозък не се увреждат. Симптомите се появяват по -често след тридесетгодишна възраст. Известни са случаи на диагностика в детска възраст. Навременното лечение дава благоприятна прогноза.
- Вторият тип е невронопатична детска форма, която се среща рядко. Симптомите се появяват в ранна детска възраст след шест месеца. Има прогресивно увреждане на мозъка на детето. Смъртта може да дойде внезапно от задушаване. Всички деца умират преди двегодишна възраст.
- Третият тип (невронопатична ювенилна форма). Симптомите се наблюдават от 10 -годишна възраст. Укрепването на знаците е постепенно. Хепатоспленомегалията - увеличен черен дроб и далак - е безболезнена и не уврежда чернодробната функция. Възможно увреждане на психологията на поведението, появата на неврологични усложнения, портална хипертония, венозно кървене и смърт. Увреждането на костната тъкан от клетките на Гоше може да доведе до ограничена подвижност и увреждане.
Симптоми на болестта на Гоше
Клиничната картина зависи от вида на заболяването, но има общи признаци на това заболяване. Можете да подозирате синдром на Гоше (вижте снимката) по следните прояви:
- бледност на кожата;
- нарушен растеж при деца;
- обща слабост;
- възпаление на лимфните възли;
- увеличен черен дроб и далак;
- фрактури на фона на отсъствие на наранявания;
- спонтанно кървене от носа;
- хеморагични звездички по кожата.
Синдромът на Гоше не зависи от пола на детето. В допълнение, симптомите на заболяването често наподобяват клиничната картина на хематологични патологии. Това затруднява диагностицирането на болестта.
Типични признаци на различни форми на синдрома на Гоше:
Болест на Гоше при деца
Симптомите могат да започнат да се проявяват на различна възраст. Вторият вид заболяване често се проявява дори на възраст от шест месеца. Пациентите в този случай живеят до 1-2 години. Третият тип е характерен за деца на възраст 2-4 години, въпреки че понякога се отбелязва в юношеска възраст. Същото важи и за първата форма. Може да се появи както в ранна детска възраст, така и в юношеска възраст. Симптомите на синдрома на Гоше при деца:
- лоша способност за смучене и преглъщане;
- нарушения на движението на очите;
- конвулсии;
- дихателни нарушения;
- магарешка кашлица;
- жълто-кафява пигментация на кожата.
Диагностика
Събиране на анамнеза и оплаквания от заболяването (изясняване на времето на появата на първите симптоми на заболяването, как те са прогресирали с течение на времето).
Заболяването може да се подозира, ако случайно се открие уголемяване на черния дроб и далака (например според ултразвук), инхибиране на хемопоетичната система (промени в кръвните изследвания: намаляване на нивото на хемоглобина, тромбоцитите, появата на атипични кръвни клетки), появата на симптоми на увреждане на костите.
На следващия етап се провеждат специални проучвания за потвърждаване на диагнозата:
- ензимен анализ - определяне нивото на ензим (глюкоцереброзидаза) в левкоцити и кожни клетки, което дава възможност да се постави диагноза с абсолютна точност;
- биохимичен кръвен тест (намалена активност на β-глюкоцереброзидаза, повишено ниво на хитотриозидаза);
- изследване на костен мозък (наличие на характерни клетки на Гоше);
- молекулярни изследвания на генно ниво (идентифициране на генетично разстройство);
- рентгенография и компютърна диагностика (ядрено -магнитен резонанс) на костите, тъй като може да има области с по -ниска плътност, които имат специфични признаци за това заболяване.

Третият вид болест на Гоше
Как се лекува болестта на Гоше?
Специализираната грижа за пациенти с първи и трети тип заболяване е насочена към премахване на симптомите и компенсиране на първичния генетичен дефект - увеличаване на количеството на липсващия ензим, увеличаване на катаболизма на гликосфинголипидите. При патология тип 2 терапевтичните мерки не са достатъчно ефективни, усилията на лекарите се свеждат до облекчаване на клиничните прояви - болка, конвулсии, дихателни нарушения.
Общата схема включва следните области:
- Ензимно -заместителна терапия... Основното лечение е доживотна ензимна заместителна терапия (ERT) с използване на рекомбинантна глюкоцереброзидаза. Ефективността е доста висока - симптомите са напълно спрени, качеството на живот на пациентите се увеличава. ERT е препоръчително за третия и първия вид заболяване. Лекарствата се прилагат интравенозно. Честите инфузии понякога причиняват възпалителни заболявания на вените (флебит).
- Терапия за намаляване на субстрата.Тази посока е нова в лечението на болестта на Гоше, относително разпространена в САЩ и европейските страни. Насочени към намаляване на скоростта на субстратно производство на гликосфинголипиди и ускоряване на катаболизма на натрупващите се макромолекули. Лекарствата са специфични инхибитори на глюкозилцерамид синтаза. Методът е показан за заболяване тип 1 с леки до умерени симптоми.
- Симптоматична терапия... С явленията на остеопороза се предписва комплексна терапия, включваща прием на съдържащи калций лекарства, витамин D и спазване на диета, обогатена с калций. Тези мерки могат да забавят загубата на костна маса, да увеличат здравината на костите и да предотвратят фрактури. За скелетни усложнения се използват аналгетици (НСПВС), антибиотична терапия. Симптомите на неврологични нарушения се контролират от антиепилептични лекарства, ноотропи, мускулни релаксанти.
Предотвратяване
Единственият метод за предотвратяване на болестта на Гоше е генетичното консултиране. Ако в семейството се роди дете с това заболяване, при последващи бременности се определя наличието на глюкоцереброзидаза в клетките на околоплодната течност. При недостиг на този ензим в плода лекарите препоръчват прекъсване на бременността.
Прогноза
При първия тип заболяване, ранна диагностика и навременно започване на заместваща терапия за болестта на Гоше е възможна положителна динамика. Вторият вид глюкоцереброзидоза е най -неблагоприятен, тъй като протича по -тежко. Болните деца обикновено не живеят повече от две години. Третата форма на болестта на Гоше, с навременна диагностика и адекватно лечение, помага за поддържане на жизнените функции на пациента. В противен случай той умира достатъчно бързо от развиващите се усложнения.
Болестта на Гоше е генетично заболяване, при което мастните вещества (липиди) се натрупват в клетките и върху някои органи. Болестта на Гоше е най -често срещаното заболяване на лизозомното съхранение. Това е една от формите на сфинголипидоза (подгрупа от заболявания на лизозомното съхранение), тъй като се проявява в дисфункционалния метаболизъм на сфинголипидите.
Разстройството се характеризира с умора, анемия, ниски кръвни тромбоцити и увеличен черен дроб и далак. Това се дължи на наследствен дефицит на ензима глюкоцереброзидаза, който действа върху глюкозилцерамид на мастни киселини. Когато ензимът е повреден, глюкозилцерамид се натрупва, по -специално в левкоцити, най -често в макрофаги (моноядрени левкоцити). Глюкозилцерамидът може да се натрупва в далака, черния дроб, бъбреците, белите дробове, мозъка и костния мозък.
Болестта на Гоше може да включва увеличен далак и черен дроб, тежки неврологични усложнения, подуване на лимфните възли и околните стави, подуване на корема, кафеникав цвят на кожата, анемия, нисък брой на тромбоцитите в кръвта и склерата.
Заболяването се причинява от рецесивна мутация в ген, разположен на хромозома 1, и засяга както мъжете, така и жените. Около 1 на 100 души по света носят болестта на Гоше. Заболяването е кръстено на френския лекар Филип Гоше, който първоначално го е описал през 1882 г.
Видове болести на Гоше
Болестта на Гоше има три общи клинични подтипа: тип I, тип II и тип III.
Тип I е най -честата форма на заболяването, с честота 1 на 50 000 новородени. Симптомите на този тип болест на Гоше могат да се появят в ранна възраст или в зряла възраст и включват:
- Уголемен черен дроб и силно увеличен далак;
- Слабост на скелетните кости;
- Анемия, тромбоцитопения и левкопения;
- Увреждане на бъбреците;
- Умора.
Тип II обикновено започва да се проявява през първите шест месеца от раждането и се среща с честота около 1 на 100 000 новородени. Симптомите на този тип болест на Гоше включват:
- Уголемяване на черния дроб и далака;
- Обширни и прогресивни мозъчни увреждания;
- Нарушения на движението на очите, спастичност, конвулсии и скованост на крайниците;
- Слаба способност за смучене и преглъщане.
Засегнатите деца обикновено умират на възраст от 2 години.
Тип III (хронична невропатична форма) може да започне по всяко време, през детството или дори в зряла възраст, и се среща с честота 1 на 100 000 новородени. Основните симптоми включват увеличен далак или черен дроб, гърчове, лоша координация, проблеми с дишането, скелетни аномалии, нарушения на движението на очите и кръвни нарушения, включително анемия.
Симптоми на болестта на Гоше
Честите симптоми на болестта на Гоше са:
- Безболезнена хепатомегалия и спленомегалия - размерът на далака може да бъде от 1500 до 3000 ml, за разлика от нормалния размер от 50-200 ml. Спленомегалията може да намали апетита чрез натиск върху корема, а уголемяването на далака увеличава риска от разкъсване на далака;
- Хиперспленизъм и панцитопения - бързо и преждевременно разрушаване на кръвни клетки, което води до анемия, неутропения, левкопения и тромбоцитопения (с повишен риск от инфекция и кървене);
- Цироза на черния дроб;
- Тежка болка в ставите и костите, често в тазобедрените и коленните стави
- Неврологични симптоми;
- Тип II: тежки гърчове, хипертония, умствена изостаналост, апнея;
- Тип III: мускулни потрепвания, гърчове, деменция, опраксиална мускулна апраксия;
- Остеопороза;
- Жълтеникавокафява пигментация на кожата.
Лечение на болестта на Гоше
 Лечението на подтипове 1 и 3 на болестта на Гоше може да започне с интравенозно заместване на рекомбинантния глюкоцереброзидазен ензим, което може значително да намали размера на черния дроб и далака, скелетните аномалии и да обърне други прояви. Тази процедура струва около 200 000 долара на пациент и трябва да се повтаря годишно през целия живот на пациента. Болестта на Гоше се лекува и с лекарството Velaglucerase Alfa, което е одобрено като алтернативно лечение от февруари 2010 г.
Лечението на подтипове 1 и 3 на болестта на Гоше може да започне с интравенозно заместване на рекомбинантния глюкоцереброзидазен ензим, което може значително да намали размера на черния дроб и далака, скелетните аномалии и да обърне други прояви. Тази процедура струва около 200 000 долара на пациент и трябва да се повтаря годишно през целия живот на пациента. Болестта на Гоше се лекува и с лекарството Velaglucerase Alfa, което е одобрено като алтернативно лечение от февруари 2010 г.
Също така, лечението на болестта на Гоше може да бъде успешна трансплантация на костен мозък, която лекува неврологични прояви на заболяването, тъй като по време на процедурата се инжектират моноцити с активна бета-глюкозидаза. Тази процедура обаче носи значителни рискове и рядко се препоръчва за болестта на Гоше.
Хирургия за отстраняване на далака (спленектомия) рядко се налага, ако пациентът е анемичен или когато увеличен орган засяга здравето на пациента. Преливане на кръв може да се извърши при пациенти със симптоми на анемия. Също така в някои случаи се налага хирургична подмяна на ставите за подобряване на мобилността и качеството на живот.
Други лечения за болестта на Гоше включват антибиотици за инфекции, антиепилептици, бисфосфонати за костни лезии и чернодробни трансплантации.
Болестта на Гоше се лекува и с перорални лекарства, които действат на молекулярно ниво. Миглустат е едно от тези лекарства и е одобрен за лечение на болестта на Гоше през 2003 г.
Видеоклип в YouTube, свързан със статията:
Днес болестта на Гоше е едно от най -често срещаните лизозомни заболявания, при което има натрупване на липиди - мастни вещества, в различни клетки и органи, което води до тяхното увреждане.
Заболяването има генетичен произход и значително влошава жизнения стандарт на пациентите.
Болестта на Гоше се проявява под формата на дисфункция на метаболитните процеси на сфинголипидите, които са отговорни за транслацията на клетъчния сигнал и клетъчното разпознаване. Нервната тъкан е особено богата на сфинголипиди, което обяснява необратимото й увреждане при тази патология.
В резултат на генетична мутация и наследяване на засегнатия ген възниква дефицит на ензима глюкоцереброзидаза, който насърчава разграждането на глюкозилцерамид на мастни киселини. В резултат на това глюкозилцерамид се натрупва в макрофаги - мононуклеарни левкоцити и засяга мястото на натрупване. Далакът, черният дроб, костният мозък и мозъкът, белите дробове могат да се превърнат в такъв резервоар за съхранение.
Болестта на Гоше засяга както мъжете, така и жените с еднаква честота. За сто души от населението поне един е носител на патологичния ген на болестта. За първи път болестта е описана от Филип Гоше, френски лекар, в края на деветнадесети век.
Моделите на наследяване на болестта на Гоше
Наблюденията и проучванията предполагат два вида наследяване на болестта. Видовете наследяване на болестта на Гоше се класифицират по произход на автозомно -доминантни и автозомно -рецесивни.
С автозомно доминантно наследяване детето получава патологията от един от родителите. Автозомно рецесивно наследяване възниква само когато засегнатият ген е наследен от двамата родители.
При болестта на Гоше се наблюдава преобладаването на втория тип наследяване.
В допълнение, видовете наследяване на болестта на Гоше са разделени на три общи подтипа с няколко отличителни черти.
Първият вид заболяване е най -често срещаното и засяга 1 на 50 000 родени деца. В този случай симптомите на болестта на Гоше могат да се проявят както в детството, така и в зряла възраст. Те включват увреждане на бъбреците, увеличен далак и черен дроб, левкопения, анемия и тромбоцитопения. Пациентите изпитват постоянна умора и слабост. Наблюдава се отслабване на скелетните кости.
Симптомите на болестта на Гоше тип II започват да се проявяват при бебетата през първата половина на годината - от 3 до 6 месеца. Развитието на втория тип се случва наполовина по -често от първия. В допълнение към увеличаването на далака и черния дроб се наблюдават значителни промени в мозъка, в резултат на което се нарушават двигателните функции на очите, спазмите и сковаността на краката и ръцете, намаляват се способностите за преглъщане и смучене. За съжаление децата с този вид заболяване трудно могат да доживеят до 2 -годишна възраст.
Хроничната невропатична форма на заболяването, определена като трети тип, протича със същата честота като предишната. Симптомите на болестта на Гоше в този случай се допълват от дихателна недостатъчност, скелетна патология и заболявания на кръвта.
Общи признаци за всички видове заболяване могат да бъдат - намаляване на нивото на тромбоцитите, анемия, кафяв оттенък на кожата, подуване на лимфните възли и стави, неврологични аномалии.
При болестта на Гоше увеличаването на далака до значителен размер намалява апетита поради натиск върху корема. По -нататъшното увеличение може да доведе до разкъсването му. Болестта провокира разрушаване на клетъчния състав на кръвта, черния дроб, умствена изостаналост, синдроми на болка.
Диагностика на болестта на Гоше
 Заболяването обикновено се диагностицира без затруднения. Но симптомите му могат да бъдат сбъркани с проява на подобни заболявания, затова е много важно да се постави точна диагноза.
Заболяването обикновено се диагностицира без затруднения. Но симптомите му могат да бъдат сбъркани с проява на подобни заболявания, затова е много важно да се постави точна диагноза.
В тази връзка диагностиката на болестта на Гоше трябва да се извърши по метода на изключване.
За да се определи точно заболяването, активността на ензима се проверява чрез специализиран кръвен тест. Ензимен тест е най -ефективен за откриване на заболяване.
Проверява се броят на левкоцитите, тромбоцитите и еритроцитите.
Ако е необходимо, се извършва ДНК анализ за определяне на генетичния произход.
В допълнение към тези форми на диагностициране на болестта на Гоше, се извършват рентгенови изследвания, компютърна томография и ядрено-магнитен резонанс, за да се определи общото състояние на тялото. За неврологични разстройства се провеждат специални тестове с помощта на специални тестове.
Лечение на болестта на Гоше
Най -ефективното и скъпо лечение за болестта на Гоше тип 1 и 3 е интравенозно заместване на рекомбинантния глюкоцереброзидазен ензим. Тази процедура може да намали обема на черния дроб и далака, както и да намали скелетните нарушения и други аномалии на тялото. Като алтернатива се използва Velaglucerase Alfa.
Трансплантацията на костен мозък се използва в неврологична клиника на заболяването. Но трансплантацията има определен риск и се препоръчва само в изключителни случаи.
Спленектомия - отстраняване на далака, също се използва само в случаи на значителна заплаха.
Ако има прогресивно кръвопреливане. Понякога се извършва хирургична подмяна на увредени стави или чернодробна трансплантация.
Лечението на болестта на Гоше с миглустат позволява молекулярно въздействие върху болестта.
Освен това при лечението се използват антибиотици, биофосфонати и антиепилептични лекарства.
RCHD (Републикански център за развитие на здравеопазването към Министерството на здравеопазването на Република Казахстан)
Версия: Клинични протоколи на Министерството на здравеопазването на Република Казахстан - 2016 г.
Други сфинголипидози (E75.2)
Сиротични болести
Главна информация
Кратко описание
Одобрен
Съвместна комисия по качеството на медицинските услуги
Министерство на здравеопазването и социалното развитие на Република Казахстан
от 29 септември 2016 г.
Протокол No11
Болест на Гоше (HD)- болест на лизозомно съхранение, полисистемно заболяване, което се основава на дефицит на ензима глюкоцереброзидаза, водещо до прогресивно увеличаване на паренхимните органи, постепенна инфилтрация на костния мозък от макрофаги, натоварени с липиди, дълбоки нарушения на хематопоезата и в малка степен част от пациентите, увреждане на централната нервна система.
Съотношението на кодовете ICD-10 и ICD-9:
Дата на разработване на протокола: 2016 година.
Потребителипротокол:общопрактикуващи лекари, педиатри, онкохематолози.
Скала за ниво на доказателства:
| А | Висококачествен мета-анализ, систематичен преглед на РКИ или големи РКИ с много ниска вероятност (++) отклонение, чиито резултати могат да бъдат обобщени за съответната популация. |
| Б | Висококачествен (++) систематичен преглед на кохортни проучвания или проучвания за контрол на случаите или висококачествени (++) кохортни или случаи на контрол на случаите с много нисък риск от пристрастия или RCT с нисък (+) риск от пристрастия, който може да бъде обобщен към съответното население ... |
| ° С | Кохортно или контролно проучване или контролирано проучване без рандомизация с нисък риск от пристрастие (+). Резултатите от които могат да бъдат обобщени към съответната популация или РКИ с много нисък или нисък риск от пристрастие (++ или+), резултатите от които не могат да бъдат разширени директно върху съответната популация. |
| д | Описание на поредица от случаи или неконтролирано изследване или експертно мнение. |
Класификация
Класификация
В съответствие с наличието и характеристиките на клиничния ход и участието на централната нервна система (ЦНС), три вида болест на Гоше:
· Невропатични (тип I).
− Азтип -бполезениГошее най-честата форма на заболяването, при която централната нервна система не е засегната (следователно този тип се нарича още невропатичен).
Симптомите са изключително разнообразни - от асимптоматични форми до тежки увреждания на органи и кости. В интервала между тези полярни клинични групи има пациенти с умерено увеличаване на далака и почти нормален кръвен състав, със или без увреждане на костите. Въпреки че този вид заболяване понякога се нарича възрастна болест на Гоше, тя може да засегне хора от всички възрасти. Колкото по -рано се появят клиничните прояви, толкова по -тежко протича болестта.
· Невронопатични (тип II иIII).
− II тип- остър невронопатичен.Болестта на Гоше тип 2 е много рядко, бързо прогресиращо заболяване, характеризиращо се с тежко увреждане на мозъка, както и на почти всички органи и системи.
По-рано наричана болест на Гоше при новородени, болестта тип 2 се характеризира с тежки неврологични нарушения през първата година от живота на детето, възникват припадъци, страбизъм, мускулна хипертоничност, умствено и физическо забавяне на развитието. Често тази форма на HD се комбинира с вродена ихтиоза. Заболяването се развива при по -малко от 1 на 100 000 новородени. Прогресивната психомоторна дегенерация завършва със смърт, обикновено свързана с дихателна недостатъчност.
− III тип (хронична невронопатична).По-рано наричана болест на Гоше от младежки тип, болестта тип 3 се характеризира с бавно прогресиращо увреждане на мозъка, както и с тежки симптоми от други органи. Този вид заболяване също е много рядко. Признаците и симптомите на болестта на Гоше тип 3 се развиват в ранна детска възраст и съответстват на тези за заболяване тип 1, с изключение на признаци на увреждане на централната нервна система. Точна диагноза е възможна само с прогресията на симптомите на невропатия, потвърдена от клинични проучвания. Пациентите с болест на Гоше тип 3, които са навършили пълнолетие, могат да живеят повече от 30 години.
Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОТО НИВО
Диагностични критерии
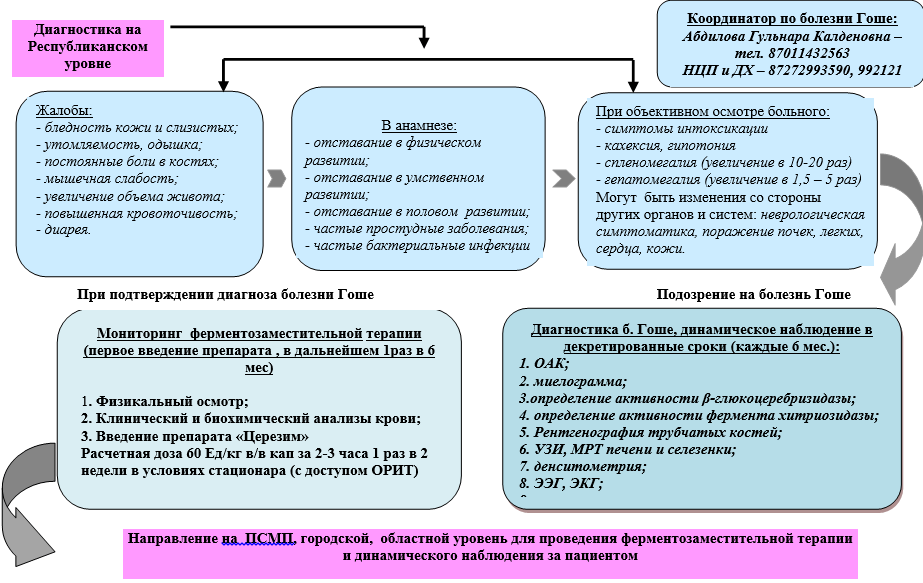
Оплаквания и анамнеза:
Слабост, повишена умора;
· Повишена чувствителност към инфекции (респираторни, бактериални);
· Прояви на хеморагичен синдром (подкожни хематоми, кървене от лигавиците) и / или продължително кървене по време на незначителни хирургични интервенции;
· Силен синдром на болка в костите и ставите (естеството и локализацията на болката, анамнеза за костни фрактури);
· Забавяне във физическото и сексуалното развитие;
· Прояви на неврологични симптоми (окуломоторна апраксия или конвергентен страбизъм, атаксия, загуба на интелигентност, нарушена чувствителност и др.);
Фамилна анамнеза (спленектомия или горните симптоми при братя и сестри, родители).
Увеличен обем на корема
Физическо изследване:
· Обща проверка;
· Измерване на ръст, телесно тегло, телесна температура;
· Оценка на състоянието на остеоартикуларната система;
· Идентифициране на признаци на хеморагичен синдром;
· Разкриване на хепатоспленомегалия, лимфаденопатия;
· Оценка на кожата в областта на колянните и лакътните стави (наличие / липса на хиперпигментация).
Клинични симптоми и признаци на болестта на Гоше в зависимост от възрастта
| Система | Симптом | Новородено |
Деца до една година |
Деца | Тийнейджъри |
| ЦНС | Забавяне и регресия на психомоторните умения | - | +++ | ++ | ± |
| конвулсии | - | +++ | ++ | ± | |
| Дермално | Колодиева кожа (подуване на гръбната част на краката и ръцете) | +++ | - | - | - |
| Стомашно-чревния тракт | Хепатоспленомегалия | ++ | +++ | +++ | +++ |
| Цироза на черния дроб | - | - | - | - | |
| Офталмологичен | Ненормални движения на очите | - | +++ | ++ | ± |
| Хематологично | анемия | - | + | +++ | ++ |
| Пяна клетки | ++ | +++ | +++ | +++ | |
| панцитопения | - | + | + | + | |
| тромбоцитопения | - | + | +++ | +++ | |
| Скелет | Болка в костите | - | - | + | +++ |
| кифоза | - | - | ± | ++ | |
| остеопороза | - | - | ± | ++ | |
| Патологични фрактури | - | - | ± | + | |
| Дихателни | Ограничаващо белодробно заболяване, белодробна хипертония | - | ++ | ++ | + |
| Други | Ранна смърт | +++ | +++ | ± | - |
| Специфични лабораторни изследвания | β-D-глюкозидаза | ↓↓↓ | ↓↓ | ↓↓ | ↓↓ |
| Хитотриозидаза |
Лабораторни изследвания :
· Удължен кръвен тест: тромбоцитопения, левкопения, анемия;
BAC: повишаването на нивото на ензимите в кръвта - ALT, AST, изследването за метаболизъм на желязото (серумно желязо, TIBS, феритин, трансферин) ще помогне при диференциалната диагноза между анемия при хронично заболяване и състояние на дефицит на желязо, което изисква стандартно лечение;
· Определяне на активността на ензима глюкоцереброзидаза и хитотриазидаза в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия - за потвърждаване на диагнозата;
· Молекулярно генетично изследване за потвърждаване на диагнозата - идентифициране на глюкоцереброзидазния ген, локализиран върху дългата ръка на хромозома 1 (регион 1q21q31);
· Морфологичното изследване на костния мозък помага да се идентифицират характерни диагностични елементи - клетки на Гоше и в същото време да се изключи диагнозата хемобластоза или лимфопролиферативна болест като причина за цитопения и хепатоспленомегалия.
Инструментални изследвания
Диагностичен алгоритъм

Алгоритъм за диагностика на болестта на Гоше при деца на градско, регионално ниво

Алгоритъм за диагностика на болестта на Гоше при деца на републиканско ниво
Диагностика (болница)
ДИАГНОСТИКА НА СТАНЦИОНАРНО НИВО
Диагностични критерии:.
Диагностичен алгоритъм


Списък на основните диагностични мерки (UD - B)
Подробен кръвен тест
· Кръвна химия
Определяне на активността на ензима глюкоцереброзидаза и хитотриазидаза
Молекулярно генетични изследвания
Ултразвук на черния дроб, далака
ЯМР на бедрените кости
ЕКГ
Рентгенова снимка на костите на скелета
Списък на допълнителните диагностични мерки:
· Миелограма - изследването на костния мозък помага да се идентифицират характерни диагностични елементи - клетки на Гоше и в същото време да се изключи диагнозата хемобластоза или лимфопролиферативна болест като причина за цитопения и хепатоспленомегалия.
· КТ на белите дробове - за изключване на патологията на белодробната система с продължителна неутропения.
· ЯМР на мозъка - за диференциална диагноза с онкологични заболявания, изключване на увреждане на ЦНС при продължителен цитопеничен синдром (риск от инсулт поради хеморагичен тип).
· ЯМР на черния дроб, далака - при наличие на хепатоспленомегалия, съществува висок риск от инфаркт на черния дроб и далака поради инфилтрацията на органи и тъкани с клетки на Гоше.
EchoCG - с тежка тахикардия, на фона на симптоми на дихателна недостатъчност с продължителен цитопеничен синдром, съществува риск от усложнения от страна на сърдечно -съдовата система (ексудативен перикардит, кардит, автономни дисфункции).
· Коагулограма-при наличие на цитопенична s-ma, възможно е добавяне на бактериална, вирусна инфекция, риск от реализиране на хеморагична s-ma, септично състояние, DIC синдром.
· Доплер ултразвук на съдовете на порталната система - за изключване на портална хипертония.
Инфекциозни усложнения на фона на продължителен цитопеничен синдром саиндикация за допълнителни лабораторни изследвания:
Бактериологично изследване на биологични течности,
Серологични (вирусологични) изследвания за CMV, хепатит B, C, (D), HIV, EBV,
Определяне на С-реактивен протеин (количествено),
С повишаване на индексите на трансаминазите: проведете серологични (вирусологични) изследвания за изключване на вирусен хепатит: CMV, A, B, C, EBV, с положителни резултати от PCR
Коагулограма - изследване на хемостаза с риск от септични усложнения, обилен хеморагичен синдром
Рентгенова снимка на костите на скелета - за идентифициране и оценка на тежестта на увреждането на остеоартикуларната система (дифузна остеопороза, характерна луковична деформация на дисталната бедрена и проксимална пищяла (колби на Erlenmeyer), огнища на остеолиза, остеосклероза и остеонекроза), патологична фрактура
· Денситометрията и ядрено -магнитен резонанс (ЯМР) са по -чувствителни методи - те позволяват диагностициране на костни лезии (остеопения, инфилтрация на костен мозък) в ранните етапи, които не са достъпни за изобразяване чрез рентгенография;
· Ултразвукът и ЯМР на черния дроб и далака могат да разкрият техните фокални лезии и да определят първоначалния обем на органите, което е необходимо за последващо проследяване на ефективността на ензимозаместителната терапия;
· Доплер ехокардиография - при спленектомирани пациенти;
езофагогастродуоденоскопия - при наличие на подходящи оплаквания или признаци на портална хипертония.
Диференциална диагноза
Диференциална диагноза
Болестта на Гоше трябва да се диференцира от всички заболявания, възникващи с хепатоспленомегалия, цитопения, кървене и увреждане на костите.
| Диагностика | Обосновка за диференциална диагноза | Анкети | Критерии за изключване на диагнозата |
| Хемобластоза и лимфоми | Хеморагичен s-m, костна болка, хепатоспленомегалия, |
2. миелограма, |
|
| Придобити апластични анемии | Хеморагичен s-m, (+ / _) костна болка, панцитопения |
1. Общ кръвен тест с преброяване на тромбоцитите, ретикулоцитите, 2. миелограма, 3. молекулярно генетичен кръвен тест |
1. Отсъствие на намаляване на активността на ензима глюкоцереброзидаза и повишаване на активността на ензима хитотриазидаза (в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия); 2. генът за глюкоцереброзидаза, локализиран върху дългото рамо на хромозома 1 (регион 1q21q31), не е идентифициран; 3. При преброяване на клетки в миелограмата не са открити клетки на Гоше |
| Хронично холестатично чернодробно заболяване, чернодробна цироза в резултат на хроничен вирусен и невирусен хепатит | Хепатоспленомегалия, повишени нива на трансаминази, билирубин, цитопеничен s-m, хеморагичен s-m, болезнен s-m |
1. Общ кръвен тест с преброяване на тромбоцитите, ретикулоцитите, 2. миелограма, 3. молекулярно генетичен кръвен тест 4. определяне на активността на ензимите глюкоцереброзидаза и хитриозидаза 5. В / х кръвен тест 6. Ултразвук, КТ, ЯМР на коремните органи |
1. Отсъствие на намаляване на активността на ензима глюкоцереброзидаза и повишаване на активността на ензима хитотриазидаза (в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия); 2. генът за глюкоцереброзидаза, локализиран върху дългото рамо на хромозома 1 (регион 1q21q31), не е идентифициран; |
| Хроничен остеомиелит, туберкулоза на костите | Осалгия, ограничаване на подвижността на крайниците |
2. миелограма, 3. молекулярно генетичен кръвен тест 4. определяне на активността на ензимите глюкоцереброзидаза и хитриозидаза 5. В / х кръвен тест |
1. Липса на признаци на цитопения (намаляване на хемоглобина, броя на тромбоцитите, левкопения), 2. Липсата на намаляване на активността на ензима глюкоцереброзидаза и повишаване на активността на ензима хитотриазидаза (в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия); 3. ген на глюкоцереброзидаза, локализиран върху дългото рамо на хромозома 1 (регион 1q21q31), не е идентифициран; 4. липса на хеморагична s-ma, 5. Характерните клаватни или колбовидни отоци на пищяла („колби на Ерленмайер“) не се определят с рентгенови лъчи. 5. Без хепатоспленомегалия |
| Други наследствени ферментопатии (болест на Ниман-Пик |
Ранно начало на развитие на заболяването (3-5 месеца), нараства обем на корема, забавено психомоторно развитие, гърчове, други неврологични симптоми, коремна болка, кървене, емоционална нестабилност |
1. Общ кръвен тест с преброяване на тромбоцити, ретикулоцити, 2. миелограма, 3. молекулярно -генетичен кръвен тест (определяне на мутации в гена SMPD1, NPC1 и NPC2, гена на глюкоцере брозидазата, локализиран на дългата ръка на хромозома 1 (регион 1q21q31). 4. определяне на активността на ензимите глюкоцереброзидаза и хитриозидаза, сфингомиелиназа 5. В / х кръвен тест 6. Ултразвук, КТ, ЯМР на коремните органи 7. Рентгеново изследване на костна тъкан (R, MRI, CT) 8. Преглед от невролог |
1. Отсъствие на намаляване на активността на ензима глюкоцереброзидаза и повишаване на активността на ензима хитотриазидаза (в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия); |
| Хистиоцитоза | Осалгия, ограничаване на подвижността на крайниците, панцитопения, хеморагичен s-m, хепатоспленомегалия, пневмония, склонност към инфекции |
1. Общ кръвен тест с преброяване на тромбоцитите, ретикулоцитите, 2. миелограма, имунофенотипиране на костния мозък 3. молекулярно генетичен кръвен тест 4. определяне на активността на ензимите глюкоцереброзидаза и хитриозидаза 5. В / х кръвен тест 6. Ултразвук, КТ, ЯМР на коремните органи 7. Рентгеново изследване на костна тъкан (R, MRI, CT) |
1. Отсъствие на намаляване на активността на ензима глюкоцереброзидаза и повишаване на активността на ензима хитотриазидаза (в сухи кръвни петна чрез тандемна масспектрометрия или флуориметрия); 2. генът за глюкоцереброзидаза, локализиран върху дългото рамо на хромозома 1 (регион 1q21q31), не е открит; 3 .. Рентгеновото изследване не определя характерното подуване на пищяла с форма на колба или колба ("колби Ерленмайер"). |
Лечение в чужбина
Подложете се на лечение в Корея, Израел, Германия, САЩ
Получете съвет относно медицинския туризъм
Лечение
Препарати (активни съставки), използвани при лечението
| Азитромицин (азитромицин) |
| Алфакалцидол (Alfakaltsidol) |
| Амфотерицин В (амфотерицин В) |
| Ацикловир |
| Ванкомицин (Vancomycin) |
| Вориконазол |
| Гентамицин |
| Диклофенак (диклофенак) |
| Ибупрофен (ибупрофен) |
| Имиглуцераза |
| Имуноглобулин G (Имуноглобулин G) |
| Йодиксанол (йодиксанол) |
| Каспофунгин |
| Клиндамицин |
| Колекалциферол (Kolekaltsiferol) |
| Лактулоза (лактулоза) |
| Лорноксикам |
| Меропенем |
| Метронидазол (Metronidazole) |
| Микафунгин |
| Осеин-хидроксиапатитен комплекс |
| Парацетамол (Paracetamol) |
| Трамадол (Tramadol) |
| Флуконазол (Fluconazole) |
| Цефотаксим (Cefotaxime) |
| Цефтазидим |
| Цефтриаксон |
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОТО НИВО
Тактика на лечение
Пациентите с всички видове (I, II, III) на болестта на Гоше се лекуват амбулаторно.
Лечение без лекарства:
· Режим - медицински и защитен през периода на цитопенична с -ма, хеморагични, костни усложнения;
· Превенция на травми, рехабилитация на хронични огнища на инфекция;
· Психологическа корекция - психотерапия, психологическа адаптация.
Медикаментозно лечение
Съвременното лечение на HD се състои в предписване на доживотна ензимна заместителна терапия (ERT) с рекомбинантна глюкоцереброзидаза, която облекчава основните клинични прояви на заболяването, подобрявайки качеството на живот на пациентите с HD и без да предизвиква изразени странични ефекти. . На всеки пациент с клинични прояви на HD (HD тип 1, HD тип 3) трябва да се предпише ERT. Дозата на лекарството трябва да се избира индивидуално в съответствие с клиничните и лабораторните параметри. Във връзка с развитието на лабораторна диагностика, при изследване на братя и сестри (братя и сестри на пробандата) могат да бъдат идентифицирани деца с HD, които нямат клинични прояви. Такива пациенти се нуждаят от наблюдение, но лечението им трябва да започне едва когато се появят симптоми на заболяването.
ERT има за цел да осигури достатъчно ензим за разграждане на отпадъците. По този начин, заместващата ензимна терапия работи чрез добавяне или заместване на липсващ или дефектен ензим при пациенти с болест на Гоше.

Списък на основните лекарства
Имиглуцераза
Патогенетичното лечение на болестта на Гоше се състои в предписване през целия живот на ензимно-заместителна терапия с рекомбинантна глюкоцереброзидаза. Началната доза на имиглуцераза за приложение при тип I GD е 30-40 единици / kg без увреждане на скелета и 60 единици / kg при наличие на увреждане на костите. При тип III при деца дозата може да достигне до 100-120 единици / kg .
Лекарството се прилага чрез интравенозно капене на интервали от 1 на всеки 2 седмици. (2 пъти месечно).
Постепенното намаляване на дозата с 10-20 единици / кг е възможно с изразена положителна динамика след 1 година лечение с БГ тип 1 без увреждане на костите и след 3-4 години с първоначално увреждане на скелета. Поддържаща терапия 15-60 единици / кг интравенозно капково в продължение на 3 часа на всеки 2 седмици, за цял живот.
Протокол за заместителна терапия с ензим на имиглуцераза

Списък на допълнителни лекарства
Парацетамол
Лорнаксикам
Диклофенак
Трамадол
Алфакалцидол
Флуканазол
Калций Dz
Остеогенон
Ацикловир
Лактулоза
Цефотаксим
Цефтазидим
Цефтриаксон
Азитромицин
Гентамицин
Йодиксанол
Меропенем
Нестероидни противовъзпалителни средства:
Парацетамол - таблетки 200 mg, 500 mg; свещи. Възрастни 500 mg 3-4 пъти дневно в продължение на 3-7 дни. Деца в размер на 60 mg / kg / ден на 3-4 дози, 3-7 дни;
Ибупрофен таблетки 200 mg, 400 mg; Деца - ибупрофен 30-40 mg / kg / ден,
Лорнаксикам - 4 mg, 8 mg филмирани таблетки. Възрастни, 8 mg 2 пъти дневно, през устата, 2 седмици; лиофилизат за приготвяне на разтвор за интравенозно и мускулно приложение, 8 mg. Възрастни, 8 mg 2 пъти дневно, i / m, 10 дни;
· Диклофенак - инжекционен разтвор 2,5% в ампули от 3 ml, таблетки от 0,05 g, таблетки ретард от 0,025; 0,05 и 0,1 g; хапчета за 0,025 г. Ректални супозитории за 0,05 и 0,1 г. Гел, крем, емулгел (1 g - 0,01 g ортофен) в епруветки. Деца 2-3 mg / kg / ден, i.m., за 1-3-5 дни. Възрастни 7 mg 2 пъти дневно, i.m., 1-3-5 дни.
Трамадол - инжекционен разтвор 50 mg / ml, ректални супозитории 0,1 g, капки -2,5 mg / капачка, капсули 50 mg. Вътре обичайната начална доза за възрастни и деца над 14 години е 50 mg (отново, при липса на ефект, след 30-60 минути). Парентерално (i / v, i / m, s / c) - 50-100 mg, ректално - 100 mg (повторно приложение на супозитории е възможно след 4-8 часа). Максималната дневна доза е 400 mg (в изключителни случаи може да се увеличи до 600 mg). За деца на възраст от 1 до 14 години, през устата (капки) или парентерално-еднократна доза от 1-2 mg / kg, максималната дневна доза е 4-8 mg / kg.
Коректори на метаболизма на костите и хрущялите:
Алфакалцидол капсули 0,5 mg. Дневната доза за възрастни варира от 0,07 μg до 20 μg, за деца 0,01-0,08 μg / kg., Дневната доза за деца е 0,01-0,08 μg / kg.
· Калций D3 - таблетки за дъвчене, съдържащи (активни съставки): калциев карбонат - 1250 mg (съответства на 500 mg елементарен калций); холекалциферол - 200 IU (международни единици). Възрастни и деца над 12 години - 2 таблетки дневно, основно по време на хранене.
Остеогенон - таблетки от осеин -хидроксиапатитен комплекс - 830mg; 2-4 раздела x 2 пъти на ден.
Алгоритъм на действие в извънредни ситуации

Хирургическа интервенция:не.
Други лечения:
· Психосоциална рехабилитация: психотерапия, психологическа адаптация, екологична терапия;
· Социална адаптация и подобряване на качеството на живот.
Показания за консултация със специалист :
| Специалист | Индикация |
| травматолог - ортопед |
Изключване на наличието на скелетна патология при дете |
| Невропатолог, психоневролог | оценка на неврологичен статус, невропсихичен статус, определяне на вида на заболяването |
| физиотерапевт |
определяне на методи за физиотерапевтично лечение |
| лекар за упражнения | избор на индивидуална програма от физиотерапевтични упражнения |
| генетик | потвърждаване на диагнозата, генотипизиране |
| При необходимост е възможна консултация с други специалисти, в зависимост от клиничния случай. | |
Превантивни действия:
· Ранна диагностика на клиничните прояви на болестта на Гоше за предотвратяване на усложнения;
· Медицинско генетично консултиране, за да се обясни генетичният риск.
· Превенцията на инфекциозни усложнения на фона на продължителен цитопеничен синдром в някои случаи е основната причина, в някои случаи дори смъртта на пациент.
· Грижа за устната кухина: 6-10 пъти на ден изплакване на устната кухина с дезинфекционни разтвори, предназначени за лечение на устната лигавица. Пълна, но нежна грижа за зъбите и венците; ограничаване на използването дори на меки четки за зъби; дайте предпочитание на устния душ; с тромбоцитопения или уязвими лигавици, използването на четки за зъби трябва да се изключи, вместо това е необходимо допълнително почистване на устата със стягащи вещества.
Когато се появят признаци на стоматит, е необходимо да се добави към основната терапия:
Флуконазол - прогнозна доза 4-5 mg / kg на ден, капсули 50 mg, 100 mg, 150 mg, инфузионни разтвори 2 mg / ml, гел за перорално лечение p, o i / v,
· Ацикловир - прогнозна доза 250 mg / m2 x 3 пъти на ден, таблетки 200 mg, инжекционен разтвор 250 mg, мехлем за външна употреба.
Ако се появят дефекти на устната лигавица: изключете използването на четки за зъби
2) с развитието на широко разпространен некротизиращ стоматит е показана системна противогъбична и антибактериална терапия:
· Цефотаксим, бутилка от 1 g за приготвяне на разтвор. Възрастни 1-2 g, 2-3 пъти на ден, IV, IM, 7-10 дни. Деца 50-100 mg / kg телесно тегло / ден, 2-4 пъти на ден, i / m, i / v, 7-10 дни;
Цефтазидим, бутилка 250 mg, 500 mg, 1 g, 2 g за приготвяне на разтвор. Възрастни: 1-6 g / ден в 2 или 3 дози интравенозно или интрамускулно. Деца над 2 месеца: 30-100 mg / kg / ден в 2-3 дози, с намален имунитет-до 150 mg / kg / ден (максимум 6 g / ден) в 3 дози. За новородени и кърмачета до 2 месеца: 25-60 mg / kg / ден в 2 разделени дози.
· Цефтриаксон, бутилка 500 mg, 1 g за приготвяне на разтвор. Деца 50-80mg / kg / ден IV капачка 1 час 7-10 дни;
Йодиксанол, инжекционен разтвор, 100 mg / 2 ml и 500 mg / 2 ml. Възрастни и деца на възраст от 12 години се предписват интрамускулно, интравенозно (струйно, в рамките на 2 минути или капково) по 5 mg / kg на всеки 8 часа или 7,5 mg / kg на всеки 12 часа в продължение на 7-10 дни.
· Гентамицин, инжекционен разтвор, 40 mg / ml ампули. възрастни 3-5 mg / kg (максимална дневна доза) в 3-4 дози, 7-10 дни. Малките деца се предписват само по здравословни причини с тежки инфекции. Максималната дневна доза за деца от всички възрасти е 5 mg / kg.
Азитромицин капсули 250, 500 mg. Деца с тегло над 10 kg в размер на: на първия ден - 10 mg / kg телесно тегло; в следващите 4 дни - 5 mg / kg. Възможен е 3-дневен курс на лечение; в този случай единичната доза е 10 mg / kg. (Основна доза 30 mg / kg телесно тегло). Възрастни с инфекции на горните и долните дихателни пътища, инфекции на кожата и меките тъкани се предписват 0,5 g на първия ден, след това 0,25 g от втория до петия ден или 0,5 g дневно в рамките на 3 дни (курсова доза 1,5 ж) ж.
· Меропенем, прах за приготвяне на разтвор за интравенозно приложение на 0,5 и 1,0 g. За деца на възраст от 3 месеца до 12 години препоръчителната доза е 10-20 mg / kg на всеки 8 часа, в зависимост от вида и тежестта на инфекцията, чувствителността на патогена и състоянието на пациента. При деца с тегло над 50 kg трябва да се използва доза за възрастни.
3) Дезактивацията на червата се извършва по избор на болницата, възможно е да се откаже от обеззаразяване. При първоначални чревни лезии се препоръчва обеззаразяване (превантивна терапия). За селективно обеззаразяване на червата:
Ципрофлоксацин в доза 20 mg / kg на ден, 100 mg във флакон, 250 mg, 500 mg в таблетки, капки за очи, капки за уши;
4) Наложително е да се спазва личната хигиена за всички, които се грижат за болните - родители и посетители, постоянно миене на ръцете.
Тактика на заместваща терапияи съгласно Заповед No 666 „За одобряване на Номенклатурата, Правила за набавяне, преработка, съхранение, продажба на кръв, както и Правила за съхранение, преливане на кръв, нейните компоненти и кръвни продукти от 6 март 2011 г., Приложение към Заповед No 417 Заповед от 29.05.2015 г. 2015 година.
Мониторинг на пациента:
· Доживотен FZT;
Динамичен контрол: 1 година - 1 път на 3 месеца, след това 1 път на 6 месеца:
· Социална адаптация;
· Наблюдение от генетик на семейството на пациент с HD.
Показатели за ефективност на лечението:
· Подобряване / стабилизиране на хематологичните параметри (облекчаване на цитопеничния синдром, липса на зависимост от кръвопреливане);
Възстановяване на нивото на глюкоцереброзидаза, намаляване на индекса на хитотриозидаза;
· Премахване на болезнените s-ma;
· Възстановяване на костната тъкан;
· Подобряване / стабилизиране на функцията на извън коремни органи (сърце, бели дробове, очи);
· Намаляване на честотата на респираторни инфекции;
· Намаляване на скоростта на прогресиране на заболяването;
подобряване на качеството на живот на пациента (възстановяване на умственото, духовното, физическото развитие).
Лечение (линейка)
ДИАГНОСТИЧНИ МЕРКИ, ПРОИЗВЕДЕНИ НА ЕТАПА НА АВАРИЙНАТА
Диагностични мерки:
· Събиране на анамнеза;
· Физическо изследване;
· Определение на сърдечна патология (пулсова оксиметрия, кръвно налягане, сърдечна честота, ЕКГ).
Медикаментозно лечение
· Сърдечно -белодробна реанимация по показания;
· Синдромно-симптоматична терапия по показания;
· Кислородна терапия;
· Предотвратяване на аспирация;
Аналгетична противовъзпалителна терапия
Лечение (болнично)
СТАНЦИОНАРНО ЛЕЧЕНИЕ
Тактика на лечение: наблюдавайте амбулаторно ниво.
Медикаментозно лечение:наблюдавайте амбулаторно ниво.
Медицинското лечение се извършва в съответствие с клиничните протоколи за хазартни усложнения.
Лекарствената терапия се засилва, когато възникнат усложнения на фона на дълготраен цитопеничен синдром, наслояване на вирусна / бактериална инфекция и прогресия на основното заболяване. Най-сериозните животозастрашаващи усложнения са инфекциозните усложнения. Наличие на треска при пациент с неутропения (неутрофили< 500/мкл) считается однократное повышение температуры тела >37.9 0 С продължителност повече от час или няколко покачвания (3-4 пъти на ден) до 38 0 C. Като се има предвид високият риск от фатален изход на инфекцията, треската при пациент с неутропения се счита за наличие на инфекция, което диктува незабавното започване на емпирична антибиотична терапия и провеждане на проучване за изясняване на естеството на инфекцията. Предложени са много първоначални антибактериални режими, чиято ефективност като цяло е идентична.
Общи разпоредби:
· При избора на начална комбинация от антибиотици е необходимо да се вземат предвид: резултатите от многократни бактериологични изследвания в тази клиника при други пациенти; продължителност на текущата неутропения, инфекциозна история на пациента, предишни курсове на антибиотици и тяхната ефективност
· Наред с появата на треска, всички други клинични данни: артериална хипотония, нестабилна хемодинамика са индикация за незабавното предписване на комбинация от антибиотици: карбопенеми (меропенем (или имипенем / циластатин)) + аминогликозид (амикацин) + ванкомицин.
· Дългогодишен CVC и треска след измиването му и / или не само треска, но огромен втрисане ® Ванкомицин вече е в началната комбинация;
· Клиника за ентероколит с диария: към първоначалната комбинация - ванкомицин per os 20 mg / kg на ден. Може би назначаването на метронидазол (per os и / или iv)
Тежък стоматит с възпалителни промени във венците ® пеницилин, клиндамицин в комбинация с бета-лактам или меропенем /
Характерен обрив и / или наличие на гъбични друзи в урината и / или характерни лезии в черния дроб и далака при сонография®
· Амфотерицин В - лиофилизат за приготвяне на разтвор. Началната доза е 0,5 mg / kg на първия ден, на следващия ден - пълната терапевтична доза е 1 mg / kg веднъж дневно. Когато се използва амфотерицин В, е необходимо да се следи бъбречната функция и да се направи биохимичен кръвен тест (електролити, креатинин). Необходима е постоянна корекция на калия до нормални стойности. По време на инфузията на Амфотерицин В, както и около 3-4 часа след инфузията, могат да се наблюдават реакции към приложението на лекарството под формата на треска, огромен втрисане, тахикардия, които се спират от аналгетици. В случай на нарушена бъбречна функция е необходимо да се използват вориконазол, Cancidas, липидни форми на амфоторецин В.
Вориконазол - 50 mg таблетка, лиофилизат за приготвяне на разтвор 200 mg / флакон SD SD 4-6 mg / kg.
Каспофунгин - лиофилизат за приготвяне на инфузионен разтвор 50 mg
Микофунгин - лиофилизат за приготвяне на инфузионен разтвор 50 mg
Промяна на антибиотици, като се вземе предвид чувствителността на изолираната флора. Ефективността на началната антибиотична терапия трябва да бъде оценена след 72 часа, но винаги е необходимо многократно подробно изследване на такъв пациент на интервали от 8-12 часа с оценка на стабилността на хемодинамиката и степента на интоксикация, външния вид на нови инфекциозни огнища. Антибиотичната терапия продължава до отшумяването на неутропенията и пълното разрешаване на всички инфекциозни огнища.
При дълбока аплазия, риск от развитие на септични усложнения, пасивна имунизация с имуноглобулини G - 0,1-0,2 g / kg / ден IV капачка.
Списък на основните лекарства:
Имиглуцераза 30-60 U / kg IV капачка 3 часа
Списък на допълнителни лекарства:
Парацетамол
Лорнаксикам
Диклофенак
Трамадол
Алфакалцидол
Флуканазол
Калций Dz
Остеогенон
Ацикловир
Лактулоза
Цефотаксим
Цефтазидим
Цефтриаксон
Азитромицин
Гентамицин
Йодиксанол
Меропенем
Имуноглобулин G
Амфотерицин В
Вориконазол
Каспофунгин
Микофунгин
Ванкомицин
Метронидазол
Клиндамицин
Хирургическа интервенция:
· Корекция на патологични костни фрактури, ставни контрактури.
Други лечения:
· Физическа рехабилитация: физиотерапия, лечебна гимнастика, масаж;
· Психосоциална рехабилитация: психотерапия, психологическа адаптация, екологична терапия.
Показания за консултация с тесни специалисти:наблюдавайте амбулаторно ниво.
Показания за прехвърляне в интензивното отделение и интензивното отделение:
· Декомпенсирано състояние на пациента;
· Обобщение на процеса с развитие на усложнения, изискващи интензивно наблюдение и терапия;
· Следоперативен период;
· Развитието на усложнения на фона на интензивна химиотерапия, изискваща интензивно лечение и наблюдение.
Показатели за ефективност на лечението:
· Възстановяване на умственото, духовното, физическото развитие;
· Възстановяване на мобилността, работоспособността;
· Премахване на болковия синдром през първите 2 години от терапията;
· Предотвратяване на костни кризи;
· Превенция на развитието на остеонекроза и субхондрални колапси;
· Подобряване на индекса на костната минерална плътност;
· Увеличаване на костната минерална плътност за 3 години терапия;
· Постигане на нормални темпове на растеж според популационните стандарти в рамките на 3 години от терапията;
· Достигане на нормалната възраст на настъпване на пубертета.
· Нормализиране на кръвната картина през първите 3 години от терапията;
· Намаляване на хепатоспленомегалията;
· Подобряване на състоянието на извън коремните органи (сърце, бели дробове, очи).
Допълнително управление:
Със стабилизиране на състоянието, възстановяване на хематологичните параметри, облекчаване на болката, интоксикация, хеморагични състояния, детето се изписва за амбулаторно лечение под наблюдението на педиатър, хематолог по местоживеене, за да продължи заместващата ензимна терапия под контролен тест . По -нататъшното наблюдение на състоянието на пациента е да се следи амбулаторното ниво.
Хоспитализация
Показания за планирана хоспитализация
Рутинната хоспитализация е показана за потвърждаване на диагнозата и за коригиране на дозата на ензимно -заместителната терапия.
Показания за спешна хоспитализация
· Цитопеничен синдром;
· Синдром на тежка болка ("костна криза");
· Патологична фрактура на костите на скелета;
· Дихателна недостатъчност.
Информация
Източници и литература
- Протоколи от заседанията на Съвместната комисия по качеството на медицинските услуги на Министерството на здравеопазването и социалното развитие на Република Казахстан, 2016 г.
- 1) Tooth N. V. "Болест на Гоше: разпространение, семиотика, качество на живот и клинично -икономическо обосноваване на ензимозаместителната терапия" резюме на д -р. Москва 2010 г. 2) Лукина Е.А. "Болест на Гоше: съвременното състояние на техниката" Руски медицински новини 2008, том XIII, № 2 стр. 51-56. 3) Белогурова М.Б. "Патогенеза, клинична картина, диагностика и лечение на болестта на Гоше." Педиатрия и детска хирургия. No 3 2010, стр. 43-48. 4) Aerts J. M., van Weely S., Broot R., et al. Патогенеза на лизозомни нарушения на съхранение, както е илюстрирано от болестта на Гоше // J. Inher. Metab. Дис. - 1993. - Т. 16. No 2. - P.288-291. 5) Beutler E., Grabowski G. A., Scriver C. R., et al. Метаболитните и молекулярни основи на наследствено заболяване // McGraw-Hill, Ню Йорк, 2001.-С. 3635-3668. 6) de Frost М., vom Dahl S., Weverling G. J., et al. Повишена честота на рак при възрастни болест на Гоше в Западна Европа // Кръвни клетки Mol. Дис. - 2006. - Т. 36.-С.53-58. 7) Taddei T.H., Kacena K.A., Yang M., et al. Непознатият прогресивен характер на болестта на Гоше на N370S и оценка на риска от рак при 403 пациенти // Am. J. Hematol. - 2009. - Т. 84. No 4. - С.208-214. 8) Болест на Niederau C. Gaucher. Бремен: UNI-MED; 2006.84 стр. 9) Zimran A., Kay A., Beutler E. et al. Болест на Гоше: клинични, лабораторни, радиологични и генетични особености на 53 пациенти. Медицина 1992; 71: 337-53. 10) Weinreb N. J. Болест на Гоше тип I при пациенти в напреднала възраст. Клиника Гоше. Персп. 1999; 7 (2): 1-8. 11) Воробьев А. И. (ред.) Рационална фармакотерапия на заболявания на кръвната система. М.: Легла, 2009, 563–6. 12) А.В. Давидов „Болести на лизозомното съхранение: болест на Гоше“ Сибирски медицински вестник, 2009 г., № 5. S.9-14. 13) Mikosch P., Reed M., Baker R., et al. Промени в костния метаболизъм при седем пациенти с болест на Гоше, лекувани последователно с имиглуцераза и миглустат // Калциф. Тъкан Int. - 2008. - Т. 83, No 1. - С.43-54. 14) vom Dahl S., Poll L., Di Rocco M., et al. Базирани на доказателства препоръки за наблюдение на костно заболяване и отговор на ензимно -заместителна терапия при пациенти с Гоше // Current Med. Изследване и мнение. - 2006. –Том. 22. No 6. - С.1045-1064. 15) Wenstrup R. J., Roca-Espiau M., Weinreb N. J., et al. Скелетни аспекти на болестта на Гоше: преглед // Br. J. Radiol. 75. - С.2-12. 16) Cox TM, Schofield JP Болест на Гоше: клинични характеристики и естествена история Клинична хематология на Bailliere. 1997; 10 (4): 657-689. 17) Грабовски Г. Болест на Гоше: Ензимология, генетика и лечение. В: Harris H, Hirshchorn K, eds. Напредък в човешката генетика. Ню Йорк, Ню Йорк: Plenum Press; 1993; 21: 377-441. 18) Воробьев А. И. (ред.) Рационална фармакотерапия на заболявания на кръвната система. М.: Легла, 2009, 563–6. 19) Панел за оценка на технологиите на NIH относно болестта на Гоше, болестта на Гоше: актуални проблеми в диагностиката и лечението, JAMA. 1996; 275: 548-553. Панел за оценка на технологиите на NIH относно болестта на Гоше, болестта на Гоше: актуални проблеми в диагностиката и лечението, JAMA. 1996; 275: 548-553. 20) Грабовски Г. А. Фенотип, диагностика и лечение на болестта на Gausher, s // Lancet. 2008. - Т. 372. # 9645.-П. 1263-1271. 21) Абдилова Г. К., Боранбаева Р. З., Омарова К. О. и др. Насоки „Съвременна диагностика и лечение на болестта на Гоше при деца в Казахстан“, Алмати 2015, стр. 26-27. 22). Ръководство на лекаря за диагностика, лечение и проследяване на наследствени метаболитни заболявания, изд. Н. Блау, М. Дюран, К.М. Гибсън, К. Диониси-Вичи. 2014) 23) "Федерални клинични насоки за предоставяне на медицинска помощ на деца с болест на Гоше" Москва, 2015 г.
Информация
СЪКРАЩЕНИЯ, ИЗПОЛЗВАНИ В ПРОТОКОЛА:
ALT - аланин аминотрансфераза
AST - аспарагинова аотоколаминотрансфераза
BG - Болест на Гоше
ЯМР - Ядрено -магнитен резонанс
KLA - пълна кръвна картина
OAM - общ анализ на урината
Ултразвук - ултразвуково изследване
ERT - ензимно -заместителна терапия
ЕКГ - електрокардиограма
Ехокардиография - ехокардиография
LPN - лизозомни болести за съхранение
ЦНС - централна нервна система
ДНК - дезоксирибонуклеинова киселина
HS - хеморагичен синдром
СУЕ - скорост на утаяване на еритроцитите
Компютърна томография
СПИСЪК НА РАЗРАБОТЧИЦИТЕ НА ПРОТОКОЛ:
1) Боранбаева Риза Зулкарнаевна - доктор на медицинските науки, директор на Научния център по педиатрия и детска хирургия.
2) Абдилова Гулнара Калденовна - кандидат на медицинските науки, заместник -директор на Научния център по педиатрия и детска хирургия на Републиканското държавно предприятие по педиатрия.
3) Омарова Кулян Омаровна - доктор на медицинските науки, професор, главен изследовател на Научния център по педиатрия и детска хирургия.
4) Манджуова Лязат Нурбапаевна - кандидат на медицинските науки, ръководител на катедрата по онкохематология за по -големи деца на Научния център по педиатрия и детска хирургия.
5) Сатбаева Елмира Маратовна - кандидат на медицинските науки, RSE в REM "Казахстански национален медицински университет на името на С. Д. Асфендияров", ръководител на катедрата по фармакология.
УКАЗАНИЕ НА БЕЗ КОНФЛИКТ НА ИНТЕРЕСИ:отсъстващ.
РЕЦЕНЗАРИ:
1. Курманбекова Сауле Каспаковна - професор от катедрата по стаж и резиденция по педиатрия No 2 на Казахския национален медицински университет. С. Д. Асфендияров.
УКАЗАНИЕ НА УСЛОВИЯТА ЗА ПРЕГЛЕД НА ПРОТОКОЛА:преразглеждане на протокола 3 години след влизането му в сила и / или когато станат достъпни нови диагностични / лечебни методи с по -високо ниво на доказателства.
Прикачени файлове
Внимание!
- Самолечението може да причини непоправима вреда на вашето здраве.
- Информацията, публикувана на уебсайта на MedElement и в мобилните приложения "MedElement", "Lekar Pro", "Dariger Pro", "Болести: Ръководство на терапевт" не може и не трябва да замества лична консултация с лекар. Не забравяйте да се свържете с доставчик на здравни услуги, ако имате някакви медицински състояния или симптоми, които ви притесняват.
- Изборът на лекарства и тяхната дозировка трябва да се обсъдят със специалист. Само лекар може да предпише правилното лекарство и неговата доза, като се вземе предвид заболяването и състоянието на тялото на пациента.
- Уебсайтът и мобилните приложения на MedElement „MedElement“, „Lekar Pro“, „Dariger Pro“, „Болести: Ръководство на терапевта“ са изключително информационни и справочни ресурси. Информацията, публикувана на този сайт, не трябва да се използва за неоторизирани промени в предписанията на лекаря.
- Редакторите на MedElement не носят отговорност за вреди за здравето или материални щети, произтичащи от използването на този сайт.
