Boala Gaucher este obișnuit să se numească o încălcare a metabolismului sfingolipidic, care este un răspuns la o deficiență a unei enzime care distruge glucocerebrazida, o astfel de complicație poate duce la depunerea glucocerebrozidului. Cele mai frecvente simptome ale bolii Gaucher sunt hepatosplenomegalia sau modificările sistemului nervos central. Pentru a diagnostica corect boala, este necesar să se efectueze studii citochimice ale leucocitelor.
Este o boală care nu apare atât de des, se transmite ereditar atunci când ambii părinți sunt purtători ai genei defecte. Prima dată când boala Gaucher a văzut lumina zilei în paginile manualelor medicale din 1882.
Lipsa enzimei beta-glucocerebrosidază în organele celulare înconjurate de membrană poate duce la formarea unei cantități mari de mediu nutritiv pentru microorganismele acestei materii organice în celulele sistemului macrofag al întregului organism, de regulă, acest proces apare și se dezvoltă în glande, precum și în celulele măduvei osoase și ale splinei.
Până în prezent, știința a stabilit trei tipuri de boală Gaucher:
- Tipul 1 se găsește cel mai adesea la persoanele care au trecut de stadiul pubertății și este, de asemenea, permanent, acest tip nu poate fi caracterizat prin prezența neuronopatiei. Boala de tipul 1 poate fi numită cea mai lentă și mai frecventă tipă în care sistemul nervos central nu va fi afectat.
- Tipul 2, în care copiii devin ținte de daune, nu este atât de frecvent în știință. Cu acest tip de boală, de regulă, neuronii sunt afectați, ceea ce implică o atrofie aproape completă a întregului sistem nervos. Cu acest diagnostic, copilul moare când este încă un copil.
- Tipul 3 în știință este denumit de obicei „juvenil”, cu acest tip simptomele procesului sunt mai puțin pronunțate, caz în care atrofia celulelor neuronale este inevitabilă. Trebuie remarcat faptul că specia 3 este, de asemenea, destul de rară. Oamenii de știință caracterizează acest tip de boală prin atașarea treptată, precum și haotică, a întregului sistem nervos la acest proces.
Faptul că boala Gaucher poate exista sub diferite forme externe, precum și în condiții în care sunt observate diferite structuri interne, confirmă diversitatea modificărilor genei glucocerebrosidazei foarte structurate pe cromozomul 1. În ciuda acestui fapt, bolile cu severitate variabilă pot fi urmărite printre un genotip dat ... Locul principal în problema puterii de transformare este dat de o creștere bruscă a numărului de macrofage din organe și țesuturi, ceea ce reprezintă un răspuns la apariția unei cantități mari de glucocerebrozid, cu toate acestea, metodele de funcționare a acestuia nu sunt cunoscut.
Zigotul Gaucher, de regulă, este similar cu un oval și are o dimensiune de aproximativ 70-80 mm în diametru, precum și un citoplasm pal. Conține două sau mai multe nuclee cu pigmentare crescută, care sunt deplasate la periferie. În mijlocul acestor nuclee se află structuri proteice filamentoase, care sunt localizate simultan unul în raport cu celălalt.
În procesul de dezvoltare a bolii, se acumulează beta-glucocerebrosid, care în cele din urmă își are originea din plasmalemele dezintegrate, tinde să devină un sediment în organele celulare înconjurate de membrană și formează asemănări alungite ale țevilor cu dimensiunea de douăzeci și uneori patruzeci de mm lungime, aceste tuburi pot fi văzute la o creștere de 2-3 mii de ori. Astfel de zigote pot fi găsite în LMC, precum și într-o tumoare a sistemului limfocitar B, deoarece, ca urmare a acestor afecțiuni, se observă un proces accelerat de schimb de beta-glucocerebrozide.
Simptomele bolii Gaucher
În condiții normale, se observă o substanță organică care distruge glucocerebrasidul, care hidrolizează glucocerebrozidele, formând în același timp glucoză și ceramide. Dacă în timpul dezvoltării organismului, au fost prezente leziuni ale materiei organice obținute la nivel genetic, atunci acest lucru poate duce la faptul că celulele încep să capteze și să digere particule solide, creând astfel zigote Gaucher. Acumularea acestor zigote în spații
în jurul vaselor din substanța creierului uman provoacă procesul de înlocuire a neuronilor morți sau înlocuiți cu celule glia. Există 3 tipuri, care diferă prin tiparul de apariție și răspândire a bolilor de diferite etiologii, activitatea materiei organice și, de asemenea, prin natura manifestărilor:
Tipul 1 se caracterizează prin cea mai mare frecvență de apariție - acest tip se găsește la 90% din populație (nu neuronopat).
Activitatea, care poate fi numită reziduală, observată în materia organică, are cea mai mare rată. Primele manifestări pot apărea în perioada de la 2 ani până la bătrânețe. Principalele simptome sunt modificările celulelor osoase, dezvoltarea lentă din punct de vedere fiziologic, activitatea întârziată în timpul pubertății, hemoragia în piele. Ultimul simptom, însoțit de hemoragii nazale, este destul de frecvent. După efectuarea unei radiografii, de regulă, medicii constată că capetele oaselor lungi au fost extinse, iar placa osoasă a cavității bucale este subțiată.
Tipul 2 se caracterizează prin cea mai mică frecvență de apariție (neuronopatic acut). Cu acest tip, se observă o scădere a activității reziduale a materiei organice. Primele semne grave pot fi detectate la o vârstă fragedă - după naștere. Principalele simptome se dezvoltă rapid tulburări neurologice: inelasticitatea, din păcate, acest tip duce în cele mai multe cazuri la moarte la vârsta de aproximativ doi ani.
Tipul 3 este între cel mai frecvent și cel mai rar (neuronopat subacut). Activitatea vitală a materiei organice, precum și severitatea bolii, sunt intermediare între tipurile 1 și 2. Primele simptome de acest tip pot fi găsite în copilărie. Manifestările clinice își pot schimba indicatorii în funcție de soi și includ, de asemenea, precum și afectarea coordonării mișcărilor (Ilia), infecția organelor și țesuturilor osoase (Nib) și a bolilor degenerative ale sistemului nervos central cu opacitate corneeană (III) . Dacă, cu acest tip, pacientul supraviețuiește etapei adolescente, în viitor el, de regulă, trăiește mult timp.

Diagnosticul bolii Gaucher
Diagnosticul acestei boli constă de obicei în studiul citochimic al leucocitelor. Tipurile, precum și transportul, sunt de obicei identificate pe baza analizei naturii mutațiilor. Zigotii Gaucher au o valoare de diagnostic.
VIDEO
Tratamentul bolii Gaucher
Pentru tipurile 1 și 3, se recomandă tratamentul de substituție cu medicamente complexe speciale care utilizează glucocerebrosidază placentară sau recombinantă; cu tratamentul de tip 2, din păcate, este inutil, mai mult, este complet necunoscut științei și medicinei. În timpul tratamentului, apare o modificare a enzimei, care se efectuează pentru transportul rapid și în timp util la organoidul celular înconjurat de o membrană. Pacienților tratați cu medicamente complexe speciale li se prescrie monitorizarea zilnică a nivelului colorantului din sânge, precum și a celulelor incolore din sânge; monitorizarea constantă a dimensiunii ficatului și splinei folosind CT sau RMN; observarea continuă a leziunilor osoase cu observarea completă a întregului sistem osos, scanare cu absorptiometrie cu raze X cu dublă energie sau RMN.
De regulă, următoarele medicamente sunt prescrise pacienților: Miglustat, care trebuie administrat în anumite doze, și anume, de trei ori pe zi, o sută de mg pe cale orală, miglu-stat - acest tip de medicament reduce concentrația de glucocerebrosid și, de asemenea, devine un fel de soluție pentru pacienții care, conform anumitor motive din motive de a nu putea fi supus unui tratament cu terapie de substituție enzimatică.
De obicei, este prescris pacienților cu anemie, precum și cu scăderea numărului de leucocite și trombocite din sânge, precum și în cazul în care splina crește, ceea ce începe să provoace disconfort.
Pentru tratamentul amănunțit al pacienților cu această boală, medicii recurg la celule stem sau celule stem, cu toate acestea, acest tip de tratament este cel mai periculos pentru sănătatea și viața pacientului, prin urmare este utilizat cât mai rar posibil.
Boala aparține bolilor de depozitare lizozomală (lipidoză glucozilceramidică).
Se caracterizează printr-o deficiență a enzimei glucocerebrosidază. Acest lucru duce la tulburări metabolice. Lipidele nu sunt descompuse în produse de reconsum; glucocerebrozidul se acumulează în celulele macrofagelor. Acestea cresc, capătă aspectul caracteristic al bulelor de săpun și se instalează în țesuturile corpului.
Sindromul Gaucher se dezvoltă: ficatul, splina, rinichii cresc, iar acumularea de glucocerebrosid în celulele țesuturilor osoase și a plămânilor le distruge structura.
Ce este?
Pe scurt, boala Gaucher este o boală genetică în care substanțele grase (lipidele) se acumulează în celule și pe unele organe. Boala Gaucher este cea mai frecventă dintre bolile de depozitare lizozomală. Este una dintre formele de sfingolipidoză (un subgrup de boli de depozitare lizozomală), deoarece se manifestă în metabolismul disfuncțional al sfingolipidelor.
Tulburarea se caracterizează prin oboseală, anemie, trombocite scăzute din sânge și ficat și splină mărite. Acest lucru se datorează unui deficit ereditar al enzimei glucocerebrosidază, care acționează asupra glucozilceramidei acizilor grași. Când enzima este deteriorată, glucozilceramida se acumulează, în special, în leucocite, cel mai adesea în macrofage (leucocite mononucleare). Glucosilceramida se poate acumula în splină, ficat, rinichi, plămâni, creier și măduvă osoasă.
Motive pentru dezvoltare
La nivel genetic, mutațiile apar la gene care sunt responsabile de producerea enzimei glucocerebrosidază. Această genă cu anomalii este localizată pe primul cromozom. Aceste mutații determină o activitate enzimatică scăzută. Astfel, există o acumulare de glucocerebrozide în macrofage.
Celulele mezenchimale, numite celule Gaucher, cresc treptat și devin hipertrofiate. Deoarece apar modificări în aceste celule și se găsesc în splină, rinichi, ficat, plămâni, creier și măduvă osoasă, ele, la rândul lor, deformează aceste organe și le perturbă funcționarea normală.
Boala Gaucher este o tulburare autozomală recesivă. Prin urmare, orice persoană poate moșteni o mutație a acestei enzime cu toate caracteristicile în același raport, atât de la tată, cât și de la mamă. Astfel, gradul bolii și severitatea acesteia vor depinde de deteriorarea genelor.
În teorie, toată lumea poate moșteni gena glucocerebrozidului cu leziuni sau complet sănătoase. Ca urmare a moștenirii unei gene cu anomalii, apare o mutație a acestei enzime, dar aceasta nu înseamnă o boală. Dar când un copil primește ambele gene afectate, atunci este diagnosticată boala Gaucher. Odată cu moștenirea unei gene afectate, copilul este considerat doar purtător al bolii, prin urmare există posibilitatea transmiterii acestei trăsături, cu patologie ereditară, către generațiile viitoare. Astfel, cu ambii părinți care au boala, un copil se poate naște cu boala Gaucher în 25% din cazuri, un copil purtător - în 50% și sănătos - în 25%.
Frecvența apariției acestei patologii ereditare în rândul raselor etnice este de 1: 50.000, dar este mult mai frecventă în rândul evreilor askenazi.
Boala Gaucher este numită și boală de acumulare din cauza lipsei unei enzime care ar trebui să îndepărteze produsele metabolice dăunătoare din corp și să nu le acumuleze. Ca urmare, aceste substanțe se colectează în macrofagele unor organe și le distrug.

Clasificarea și tipurile de boală
Natura evoluției bolii este de severitate variabilă. Complicațiile apar în copilărie și la vârsta adultă. Există trei tipuri de boli:
- Primul tip non-neuronopatic. Sociologia arată că este frecventă în rândul evreilor askenazi. Acest model se numește reacția Gaucher. Tabloul clinic se caracterizează printr-un curs moderat, uneori asimptomatic al bolii. Psihologia comportamentului nu se schimbă, creierul și măduva spinării nu sunt deteriorate. Simptomele apar mai des după vârsta de treizeci de ani. Există cazuri cunoscute de diagnostic în copilărie. Tratamentul la timp oferă un prognostic favorabil.
- Al doilea tip este o formă infantilă neuronopatică, care este rară. Simptomele apar în copilărie cu șase luni. Există o afectare progresivă a creierului copilului. Moartea poate proveni brusc din sufocare. Toți copiii mor înainte de vârsta de doi ani.
- Al treilea tip (formă juvenilă neuronopatică). Simptomele au fost observate de la vârsta de 10 ani. Întărirea semnelor este treptată. Hepatosplenomegalia - ficatul și splina mărite - este nedureroasă și nu afectează funcția ficatului. Posibila afectare a psihologiei comportamentului, debutul complicațiilor neurologice, hipertensiune portală, sângerări venoase și deces. Deteriorarea țesutului osos de către celulele Gaucher poate duce la mobilitate limitată și handicap.
Simptomele bolii Gaucher
Tabloul clinic depinde de tipul bolii, dar există semne generale ale acestei boli. Puteți suspecta sindromul Gaucher (a se vedea fotografia) prin următoarele manifestări:
- paloarea pielii;
- creșterea afectată la copii;
- slăbiciune generală;
- inflamația ganglionilor limfatici;
- ficat și splină mărite;
- fracturi pe fundalul absenței leziunilor;
- sângerări nazale spontane;
- asteriscuri hemoragice pe piele.
Sindromul Gaucher nu depinde de sexul copilului. În plus, simptomele bolii seamănă adesea cu tabloul clinic al patologiilor hematologice. Acest lucru face dificilă diagnosticarea bolii.
Semne tipice ale diferitelor forme de sindrom Gaucher:
Boala Gaucher la copii
Simptomele pot începe să apară la diferite vârste. Al doilea tip de boală se manifestă adesea chiar și la vârsta de șase luni. Pacienții în acest caz trăiesc până la 1-2 ani. Al treilea tip este tipic pentru copiii de 2-4 ani, deși uneori este remarcat în adolescență. Același lucru este valabil și pentru prima formă. Poate apărea atât în copilăria timpurie, cât și în adolescență. Simptomele sindromului Gaucher la copii:
- capacitate slabă de a suge și de a înghiți;
- tulburări de mișcare a ochilor;
- convulsii;
- tulburări de respirație;
- tuse convulsivă;
- pigmentare galben-maroie a pielii.
Diagnostic
Colectarea anamnezei și a plângerilor bolii (clarificarea momentului apariției primelor simptome ale bolii, modul în care au progresat în timp).
Boala poate fi suspectată dacă un ficat mărit și splina sunt detectate accidental (de exemplu, conform ultrasunetelor), inhibarea sistemului hematopoietic (modificări ale testelor de sânge: scăderea nivelului de hemoglobină, trombocite, apariția celulelor sanguine atipice ), apariția simptomelor de afectare osoasă.
În etapa următoare, se efectuează studii speciale pentru confirmarea diagnosticului:
- analiza enzimatică - determinarea nivelului unei enzime (glucocerebrosidaza) în leucocite și celule cutanate, ceea ce face posibilă stabilirea unui diagnostic cu o acuratețe absolută;
- test de sânge biochimic (activitate scăzută a β-glucocerebrosidazei, nivel crescut de chitotriosidază);
- examinarea măduvei osoase (prezența celulelor Gaucher caracteristice);
- studii moleculare la nivelul genelor (identificarea unei tulburări genetice);
- radiografie și diagnosticare computerizată (imagistica prin rezonanță magnetică) a oaselor, deoarece pot exista zone cu o densitate mai mică care prezintă semne specifice pentru această boală.

Al treilea tip de boală Gaucher
Cum este tratată boala Gaucher?
Îngrijirea specializată pentru pacienții cu primul și al treilea tip de boală are ca scop eliminarea simptomelor și compensarea defectului genetic primar - creșterea cantității de enzimă lipsă, creșterea catabolismului glicosfingolipidelor. Cu patologia de tip 2, măsurile terapeutice nu sunt suficient de eficiente, eforturile medicilor se reduc la ameliorarea manifestărilor clinice - durere, convulsii, tulburări respiratorii.
Schema generală include următoarele domenii:
- Terapia de substituție enzimatică... Tratamentul principal este terapia de substituție enzimatică pe tot parcursul vieții (ERT) utilizând glucocerebrosidază recombinantă. Eficacitatea este destul de ridicată - simptomele sunt complet oprite, crește calitatea vieții pacienților. ERT este recomandabil pentru al treilea și primul tip de boală. Medicamentele se administrează intravenos. Infuziile frecvente cauzează uneori boli inflamatorii ale venelor (flebită).
- Terapia de reducere a substratului. Această direcție este nouă în tratamentul bolii Gaucher, relativ răspândită în Statele Unite și țările europene. Destinat reducerii ratei de producție a substratului de glicozifingolipide și accelerării catabolismului macromoleculelor acumulate. Medicamentele sunt inhibitori specifici ai glucozilceramidei sintaza. Metoda este indicată pentru boala de tip 1 cu simptome ușoare până la moderate.
- Terapia simptomatică... Odată cu fenomenele de osteoporoză, este prescrisă o terapie complexă, inclusiv aportul de medicamente care conțin calciu, vitamina D și aderarea la o dietă îmbogățită cu calciu. Aceste măsuri pot încetini pierderea osoasă, pot crește rezistența osoasă și pot preveni fracturile. Pentru complicațiile scheletice, se utilizează analgezice (AINS), antibioterapie. Simptomele tulburărilor neurologice sunt controlate de medicamente antiepileptice, nootropice, relaxante musculare.
Prevenirea
Singura metodă de prevenire a bolii Gaucher este consilierea genetică. Dacă un copil cu această boală se naște în familie, în sarcinile ulterioare, se determină prezența glucocerebrosidazei în celulele lichidului amniotic. Cu o deficiență a acestei enzime la făt, medicii recomandă întreruperea sarcinii.
Prognoza
Cu primul tip de boală, diagnosticul precoce și inițierea în timp util a terapiei de substituție pentru boala Gaucher, sunt posibile dinamici pozitive. Al doilea tip de glucocerebrozidoză este cel mai nefavorabil, deoarece se desfășoară mai sever. Copiii bolnavi de obicei nu trăiesc mai mult de doi ani. A treia formă a bolii Gaucher, cu diagnostic în timp util și tratament adecvat, ajută la menținerea funcțiilor vitale ale pacientului. În caz contrar, el moare suficient de repede din cauza complicațiilor în curs de dezvoltare.
Boala Gaucher este o tulburare genetică în care substanțele grase (lipidele) se acumulează în celule și pe unele organe. Boala Gaucher este cea mai frecventă dintre bolile de depozitare lizozomală. Este una dintre formele de sfingolipidoză (un subgrup de boli de depozitare lizozomală), deoarece se manifestă în metabolismul disfuncțional al sfingolipidelor.
Tulburarea se caracterizează prin oboseală, anemie, trombocite scăzute din sânge și ficat și splină mărite. Acest lucru se datorează unui deficit ereditar al enzimei glucocerebrosidază, care acționează asupra glucozilceramidei acizilor grași. Când enzima este deteriorată, glucozilceramida se acumulează, în special, în leucocite, cel mai adesea în macrofage (leucocite mononucleare). Glucosilceramida se poate acumula în splină, ficat, rinichi, plămâni, creier și măduvă osoasă.
Manifestările bolii Gaucher includ splina și ficatul mărite, complicații neurologice severe, umflarea ganglionilor limfatici și a articulațiilor adiacente, balonare, culoarea maronii a pielii, anemie și număr scăzut de trombocite în sânge și sclera.
Boala este cauzată de o mutație recesivă într-o genă localizată pe cromozomul 1 și afectează atât bărbații, cât și femeile. Aproximativ 1 din 100 de persoane din întreaga lume poartă boala Gaucher. Boala poartă numele medicului francez Philippe Gaucher, care a descris-o inițial în 1882.
Tipuri de boală Gaucher
Boala Gaucher are trei subtipuri clinice generale: tip I, tip II și tip III.
Tipul I este cea mai frecventă formă a bolii, cu o incidență de 1 din 50.000 de nou-născuți. Simptomele acestui tip de boală Gaucher pot apărea la începutul vieții sau la vârsta adultă și includ:
- Ficatul mărit și splina foarte mărită;
- Slăbiciunea oaselor scheletice;
- Anemie, trombocitopenie și leucopenie;
- Afectarea rinichilor;
- Oboseală.
Tipul II începe de obicei să se dezvolte în primele șase luni de la naștere și apare cu o incidență de aproximativ 1 din 100.000 de nou-născuți. Simptomele acestui tip de boală Gaucher includ:
- Mărirea ficatului și a splinei;
- Leziuni cerebrale extinse și progresive;
- Tulburări ale mișcării ochilor, spasticitate, convulsii și rigiditate a membrelor;
- Abilitate slabă de a suge și de a înghiți.
Copiii afectați mor de obicei la vârsta de 2 ani.
Tipul III (forma neuropatică cronică) poate începe în orice moment, în timpul copilăriei sau chiar la maturitate și apare cu o incidență de 1 din 100.000 de nou-născuți. Simptomele majore includ splina sau ficatul mărit, convulsii, coordonare slabă, probleme de respirație, anomalii ale scheletului, tulburări ale mișcării ochilor și tulburări ale sângelui, inclusiv anemie.
Simptomele bolii Gaucher
Simptomele frecvente ale bolii Gaucher sunt:
- Hepatomegalie și splenomegalie nedureroase - dimensiunea splinei poate fi de la 1500 la 3000 ml, spre deosebire de dimensiunea normală de 50-200 ml. Splenomegalia poate reduce pofta de mâncare prin presiunea asupra abdomenului, iar mărirea splinei crește riscul de rupere a splinei;
- Hipersplenism și pancitopenie - distrugerea rapidă și prematură a celulelor sanguine, rezultând anemie, neutropenie, leucopenie și trombocitopenie (cu un risc crescut de infecții și sângerări);
- Ciroza ficatului;
- Dureri articulare și osoase severe, adesea în articulațiile șoldului și genunchiului
- Simptome neurologice;
- Tipul II: convulsii severe, hipertensiune, retard mental, apnee;
- Tipul III: zvâcniri musculare, convulsii, demență, apraxie musculară oculară;
- Osteoporoza;
- Pigmentare a pielii maro gălbuie.
Tratamentul bolii Gaucher
 Tratamentul pentru subtipurile 1 și 3 ale bolii Gaucher poate începe cu înlocuirea intravenoasă a enzimei recombinante glucocerebrosidază, care poate reduce semnificativ dimensiunea ficatului și splinei, anomalii scheletice și inversa alte manifestări. Această procedură costă aproximativ 200.000 de dolari pe pacient și trebuie repetată anual de-a lungul vieții pacientului. Boala Gaucher este, de asemenea, tratată cu medicamentul Velaglucerase Alfa, care a fost aprobat ca tratament alternativ din februarie 2010.
Tratamentul pentru subtipurile 1 și 3 ale bolii Gaucher poate începe cu înlocuirea intravenoasă a enzimei recombinante glucocerebrosidază, care poate reduce semnificativ dimensiunea ficatului și splinei, anomalii scheletice și inversa alte manifestări. Această procedură costă aproximativ 200.000 de dolari pe pacient și trebuie repetată anual de-a lungul vieții pacientului. Boala Gaucher este, de asemenea, tratată cu medicamentul Velaglucerase Alfa, care a fost aprobat ca tratament alternativ din februarie 2010.
De asemenea, tratamentul bolii Gaucher poate fi un transplant de măduvă osoasă de succes, care tratează manifestările non-neurologice ale bolii, deoarece monocitele cu beta-glucozidază activă sunt injectate în timpul procedurii. Cu toate acestea, această procedură prezintă riscuri semnificative și este rareori recomandată pentru boala Gaucher.
Intervenția chirurgicală pentru îndepărtarea splinei (splenectomie) este rareori necesară dacă pacientul este anemic sau când un organ mărit afectează sănătatea pacientului. Transfuzia de sânge poate avea loc la pacienții cu simptome de anemie. De asemenea, în unele cazuri, este necesară înlocuirea chirurgicală a articulațiilor pentru a îmbunătăți mobilitatea și calitatea vieții.
Alte tratamente pentru boala Gaucher includ antibiotice pentru infecții, antiepileptice, bifosfonați pentru leziuni osoase și transplanturi de ficat.
Boala Gaucher este, de asemenea, tratată cu medicamente orale care acționează la nivel molecular. Miglustat este unul dintre aceste medicamente și a fost aprobat pentru tratamentul bolii Gaucher în 2003.
Videoclip YouTube legat de articol:
Astăzi, boala Gaucher este una dintre cele mai frecvente boli lizozomale, în care există o acumulare de lipide - substanțe grase, în diferite celule și organe, ceea ce duce la deteriorarea lor.
Boala are o origine genetică și afectează semnificativ nivelul de trai al pacienților.
Boala Gaucher se manifestă sub forma disfuncției proceselor metabolice ale sfingolipidelor, care sunt responsabile de traducerea semnalului celular și de recunoașterea celulară. Țesutul nervos este deosebit de bogat în sfingolipide, ceea ce explică daunele sale ireversibile în această patologie.
Ca urmare a mutației genetice și a moștenirii genei afectate, apare o deficiență a enzimei glucocerebrosidază, care favorizează descompunerea glucozilceramidei acizilor grași. Ca urmare, glucozilceramida se acumulează în macrofage - leucocite mononucleare și afectează locul acumulării. Splina, ficatul, măduva osoasă și creierul, plămânii pot deveni un astfel de rezervor de stocare.
Boala Gaucher afectează atât bărbații, cât și femeile, cu aceeași frecvență. Pentru o sută de oameni din populație, cel puțin unul este purtătorul genei patologice a bolii. Boala a fost descrisă pentru prima dată de Philippe Gaucher, medic francez, la sfârșitul secolului al XIX-lea.
Modele de moștenire a bolii Gaucher
Observațiile și studiile sugerează două tipuri de moștenire a bolii. Tipurile de moștenire a bolii Gaucher sunt clasificate după origine în tipuri autosomale dominante și autosomale recesive.
Cu moștenirea autosomală dominantă, copilul primește patologia de la unul dintre părinți. Moștenirea autosomală recesivă apare numai atunci când gena afectată este moștenită de la ambii părinți.
În boala Gaucher se observă predominanța celui de-al doilea tip de moștenire.
În plus, tipurile de moștenire a bolii Gaucher sunt clasificate în trei subtipuri generale cu mai multe trăsături distinctive.
Primul tip de boală este cel mai frecvent și afectează 1 din 50.000 de copii născuți. În acest caz, simptomele bolii Gaucher se pot manifesta atât în copilărie, cât și la vârsta adultă. Acestea includ afectarea rinichilor, splina și ficatul mărite, leucopenia, anemia și trombocitopenia. Pacienții au oboseală și slăbiciune constante. Se observă slăbirea oaselor scheletice.
Simptomele bolii Gaucher de tip II încep să apară la copii în prima jumătate a anului - de la 3 la 6 luni. Dezvoltarea celui de-al doilea tip are loc la jumătate de frecvență ca primul. În plus față de mărirea splinei și a ficatului, se observă modificări semnificative în creier, ca urmare a faptului că funcțiile motorii ale ochilor, crampele și rigiditatea picioarelor și brațelor sunt afectate, abilitățile de înghițire și supt sunt reduse. Din păcate, copiii cu acest tip de boală cu greu pot trăi până la 2 ani.
Forma neuropatică cronică a bolii, definită ca al treilea tip, apare cu aceeași frecvență ca și cea anterioară. Simptomele bolii Gaucher în acest caz sunt completate de insuficiență respiratorie, patologie scheletică și boli de sânge.
Semnele frecvente pentru toate tipurile de boală pot fi - o scădere a nivelului de trombocite, anemie, o nuanță maro a pielii, umflarea ganglionilor limfatici și a articulațiilor, anomalii neurologice.
În boala Gaucher, mărirea splinei la o dimensiune semnificativă reduce pofta de mâncare din cauza presiunii asupra abdomenului. O creștere suplimentară poate duce la ruperea acesteia. Boala provoacă distrugerea compoziției celulare a sângelui, ficatului, retard mental, sindroame de durere.
Diagnosticul bolii Gaucher
 Boala este de obicei diagnosticată fără dificultate. Dar simptomele sale pot fi confundate cu o manifestare a unor boli similare, deci este foarte important să puneți un diagnostic precis.
Boala este de obicei diagnosticată fără dificultate. Dar simptomele sale pot fi confundate cu o manifestare a unor boli similare, deci este foarte important să puneți un diagnostic precis.
În acest sens, diagnosticul bolii Gaucher ar trebui efectuat prin metoda de excludere.
Pentru a determina cu precizie boala, activitatea enzimei este verificată printr-un test de sânge specializat. Un test enzimatic este cel mai eficient în detectarea bolilor.
Se verifică numărul de leucocite, trombocite și eritrocite.
Dacă este necesar, se efectuează analiza ADN pentru a determina fundalul genetic.
Pe lângă aceste forme de diagnostic al bolii Gaucher, se efectuează examinări cu raze X, tomografie computerizată și examinare prin rezonanță magnetică nucleară pentru a determina starea generală a corpului. Pentru tulburările neurologice, testarea specială se efectuează folosind teste speciale.
Tratamentul bolii Gaucher
Cel mai eficient și mai costisitor tratament pentru boala Gaucher de tip 1 și 3 este înlocuirea intravenoasă a enzimei recombinante glucocerebrosidază. Această procedură poate reduce volumul ficatului și splinei și poate reduce tulburările scheletice și alte anomalii ale corpului. Alternativ, se utilizează Velaglucerase Alfa.
Transplantul de măduvă osoasă este utilizat într-o clinică non-neurologică a bolii. Dar transplantul are un anumit risc și este recomandat numai în cazuri excepționale.
Splenectomia - îndepărtarea splinei, este utilizată și în cazurile de amenințare semnificativă.
Dacă există o transfuzie progresivă de sânge. Uneori se face înlocuirea chirurgicală a articulațiilor deteriorate sau transplantul de ficat.
Tratamentul bolii Gaucher cu miglustat permite acțiune moleculară asupra bolii.
În plus, în tratament sunt utilizate antibiotice, biofosfonați și medicamente antiepileptice.
RCHD (Centrul Republican pentru Dezvoltarea Sănătății din Ministerul Sănătății din Republica Kazahstan)
Versiune: Protocoale clinice ale Ministerului Sănătății al Republicii Kazahstan - 2016
Alte sfingolipidoze (E75.2)
Boli orfane
Informații generale
Scurta descriere
Aprobat
Comisia mixtă pentru calitatea serviciilor medicale
Ministerul Sănătății și Dezvoltării Sociale din Republica Kazahstan
din 29 septembrie 2016
Protocolul nr. 11
Boala Gaucher (GD)- boala de stocare lizozomală, o boală polisistemică, care se bazează pe o deficiență a enzimei glucocerebrosidază, care duce la o creștere progresivă a organelor parenchimatoase, infiltrarea treptată a măduvei osoase prin macrofage încărcate cu lipide, tulburări profunde ale hematopoiezei și într-o mică parte a pacienților, afectarea sistemului nervos central.
Raportul dintre codurile ICD-10 și ICD-9:
Data elaborării protocolului: 2016 an.
Membriprotocol: medici generaliști, pediatri, oncohematologi.
Scala nivelului probelor:
| A | Meta-analiză de înaltă calitate, revizuirea sistematică a ECA sau ECA mari cu probabilitate foarte mică (++) de prejudecată ale cărei rezultate pot fi generalizate la populația relevantă. |
| B | Revizuirea sistematică de înaltă calitate (++) a studiilor de cohortă sau de control de caz sau de studii de cohortă sau de control de caz de înaltă calitate (++) cu risc foarte scăzut de prejudecată sau ECR cu risc scăzut (+) de prejudecată care poate fi generalizat populației relevante ... |
| C | Un studiu de cohortă sau caz-control sau studiu controlat fără randomizare cu risc scăzut de prejudecată (+). Rezultatele cărora pot fi generalizate la populația relevantă sau ECA cu risc foarte mic sau scăzut de prejudecată (++ sau +), ale căror rezultate nu pot fi extinse direct la populația relevantă. |
| D | Descrierea unei serii de cazuri sau cercetări necontrolate sau opinii ale experților. |
Clasificare
Clasificare
În conformitate cu prezența și caracteristicile cursului clinic și implicarea sistemului nervos central (SNC), trei tipuri de boală Gaucher:
· Non-neuropatic (tip I).
− Eutip de -butilșiGaucher este cea mai frecventă formă a bolii în care sistemul nervos central nu este afectat (prin urmare, acest tip se mai numește și neuropatic).
Simptomele sunt extrem de diverse - de la forme asimptomatice la leziuni severe ale organelor și oaselor. În intervalul dintre aceste grupuri clinice polare, există pacienți cu mărire moderată a splinei și compoziție sanguină aproape normală, cu sau fără leziuni osoase. Deși acest tip de boală este uneori numită boală Gaucher pentru adulți, aceasta poate afecta persoanele de toate vârstele. Cu cât manifestările clinice apar mai devreme, cu atât boala progresează mai severă.
· Neuronopatic (tip II șiIII).
− II tip de- neuronopatic acut. Boala Gaucher de tip 2 este o boală foarte rară, care progresează rapid, caracterizată prin leziuni severe la nivelul creierului, precum și la aproape toate organele și sistemele.
Denumită anterior boala Gaucher la nou-născuți, boala de tip 2 se caracterizează prin tulburări neurologice severe în primul an de viață al copilului, episoadele, strabismul, hipertonia musculară și întârzierile mentale și fizice ale dezvoltării. Adesea, această formă de HD este combinată cu ihtioza congenitală. Boala se dezvoltă la mai puțin de 1 din 100.000 de nou-născuți. Degenerarea psihomotorie progresivă se încheie cu moartea, de obicei asociată cu insuficiența respiratorie.
− III tip de (neuronopatic cronic). Denumită anterior boala Gaucher de tip juvenil, boala de tip 3 se caracterizează prin leziuni cerebrale progresive lent, precum și simptome severe de la alte organe. Acest tip de boală este, de asemenea, foarte rar. Semnele și simptomele bolii Gaucher de tip 3 se dezvoltă în copilăria timpurie și corespund cu cele ale bolii de tip 1, cu excepția semnelor de afectare a sistemului nervos central. Un diagnostic precis este posibil numai cu progresia simptomelor neuropatiei, confirmată de studii clinice. Pacienții cu boala Gaucher de tip 3 care au atins vârsta majoratului pot trăi mai mult de 30 de ani.
Diagnostic (ambulatoriu)
DIAGNOSTIC LA NIVELUL AMBULATOR
Criterii de diagnostic
Plângeri și anamneză:
Slăbiciune, oboseală crescută;
· Sensibilitate crescută la infecții (infecții respiratorii, bacteriene);
· Manifestări ale sindromului hemoragic (hematoame subcutanate, sângerări ale membranelor mucoase) și / sau sângerări prelungite în timpul intervențiilor chirurgicale minore;
Durere severă la nivelul oaselor și articulațiilor (natura și localizarea durerii, antecedente de fracturi osoase);
· Întârziere în dezvoltarea fizică și sexuală;
· Manifestări ale simptomelor neurologice (apraxia oculomotorie sau strabism convergent, ataxie, pierderea inteligenței, afectarea sensibilității etc.);
Antecedente familiale (splenectomie sau simptomele de mai sus la frați, părinți).
Creșterea volumului abdominal
Examinare fizică:
· Inspecție generală;
· Măsurarea înălțimii, greutății corporale, temperatura corpului;
· Evaluarea stării sistemului osteoarticular;
· Identificarea semnelor sindromului hemoragic;
· Dezvăluirea hepatosplenomegaliei, limfadenopatiei;
· Evaluarea pielii în zona articulațiilor genunchiului și cotului (prezența / absența hiperpigmentării).
Simptome clinice și semne ale bolii Gaucher în funcție de vârstă
| Sistem | Simptom | Nou nascut |
Copii până la un an |
Copii | Adolescenți |
| SNC | Întârzierea și regresia abilităților psihomotorii | - | +++ | ++ | ± |
| convulsii | - | +++ | ++ | ± | |
| Dermic | Piele de colodion (umflarea dorsului picioarelor și mâinilor) | +++ | - | - | - |
| Tract gastrointestinal | Hepatosplenomegalie | ++ | +++ | +++ | +++ |
| Ciroza ficatului | - | - | - | - | |
| Oftalmic | Mișcări anormale ale ochilor | - | +++ | ++ | ± |
| Hematologic | anemie | - | + | +++ | ++ |
| Celule de spumă | ++ | +++ | +++ | +++ | |
| pancitopenie | - | + | + | + | |
| trombocitopenie | - | + | +++ | +++ | |
| Scheletal | Durerea osoasă | - | - | + | +++ |
| cifoză | - | - | ± | ++ | |
| osteoporoză | - | - | ± | ++ | |
| Fracturi patologice | - | - | ± | + | |
| Respirator | Boală pulmonară restrictivă, hipertensiune pulmonară | - | ++ | ++ | + |
| Alte | Moarte prematura | +++ | +++ | ± | - |
| Teste de laborator specifice | β-D-glucozidaza | ↓↓↓ | ↓↓ | ↓↓ | ↓↓ |
| Chitotriosidaza |
Cercetări de laborator :
· Test de sânge extins: trombocitopenie, leucopenie, anemie;
BAC: o creștere a nivelului de enzime din sânge - ALT, AST, examinarea metabolismului fierului (fier seric, TIBS, feritină, transferină) va ajuta la diagnosticul diferențial între anemia unei boli cronice și o stare de deficit de fier care necesită tratament standard;
· Determinarea activității enzimei glucocerebrosidază și chitotriazidază în pete de sânge uscate prin spectrometrie de masă tandem sau fluorimetrie - pentru confirmarea diagnosticului;
· Cercetare genetică moleculară pentru confirmarea diagnosticului - identificarea genei glucocerebrosidazei localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31);
· Examenul morfologic al măduvei osoase ajută la identificarea elementelor de diagnostic caracteristice - celulele Gaucher și, în același timp, la excluderea diagnosticului de hemoblastoză sau boală limfoproliferativă ca fiind cauza citopeniei și hepatosplenomegaliei.
Cercetare instrumentală
Algoritm de diagnosticare

Algoritm pentru diagnosticul bolii Gaucher la copii la nivel local, regional

Algoritm pentru diagnosticul bolii Gaucher la copii la nivel republican
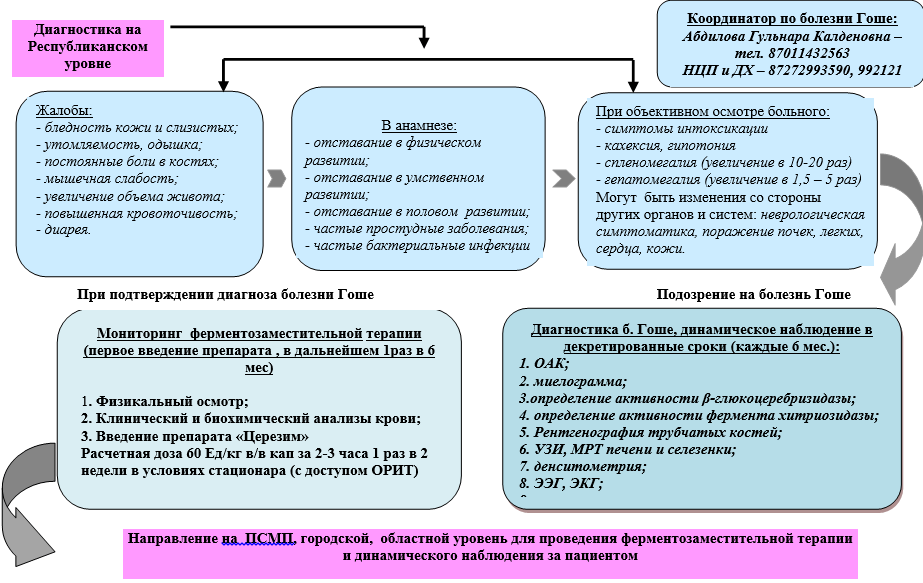
Diagnostic (spital)
DIAGNOSTIC LA NIVEL STAȚIONAR
Criterii de diagnostic:.
Algoritm de diagnosticare


Lista măsurilor diagnostice de bază (UD - B)
Test de sânge detaliat
· Chimia sângelui
Determinarea activității enzimei glucocerebrosidază și chitotriazidază
Cercetare genetică moleculară
Ecografia ficatului, splinei
RMN al oaselor coapsei
ECG
Radiografia oaselor scheletului
Lista măsurilor de diagnostic suplimentare:
· Mielogramă - un studiu al măduvei osoase ajută la identificarea elementelor diagnostice caracteristice - celulele Gaucher și, în același timp, la excluderea diagnosticului de hemoblastoză sau de boală limfoproliferativă ca fiind cauza citopeniei și hepatosplenomegaliei.
· CT plămânilor - pentru a exclude patologia sistemului pulmonar cu neutropenie prelungită.
· RMN cerebral - pentru diagnostic diferențial cu boli oncologice, excluderea afectării SNC în caz de sindrom citopenic prelungit (risc de accident vascular cerebral de tip hemoragic).
· RMN al ficatului, splinei - în prezența hepatosplenomegaliei, există un risc ridicat de infarct al ficatului și splinei datorită infiltrării organelor și țesuturilor cu celule Gaucher.
EchoCG - cu tahicardie severă, pe fondul simptomelor insuficienței respiratorii cu sindrom citopenic prelungit, există riscul de complicații ale sistemului cardiovascular (pericardită exudativă, cardită, disfuncții autonome).
· Coagulogramă - în prezența s-ma citopenică, este posibilă adăugarea unei infecții bacteriene, virale, riscul de realizare a s-ma hemoragic, stare septică, sindrom DIC.
· Ecografia Doppler a vaselor sistemului portal - pentru a exclude hipertensiunea portală.
Complicațiile infecțioase pe fondul sindromului citopenic prelungit suntindicație pentru teste suplimentare de laborator:
Examenul bacteriologic al fluidelor biologice,
Studii serologice (virologice) pentru CMV, Hepatita B, C, (D), HIV, EBV,
Determinarea proteinei C reactive (cantitative),
Cu o creștere a indicilor transaminazelor: efectuați studii serologice (virologice) pentru a exclude hepatita virală: CMV, A, B, C, EBV, cu rezultate PCR pozitive
Coagulogramă - un studiu al hemostazei cu risc de complicații septice, sindrom hemoragic abundent
Raze X ale oaselor scheletului - pentru a identifica și evalua severitatea leziunilor sistemului osteoarticular (osteoporoză difuză, deformare bulboasă caracteristică a tibiei distale femurale și proximale (baloane Erlenmeyer), focare de osteoliză, osteoscleroză și osteonecroză), fractură patologică
· Densitometria și imagistica prin rezonanță magnetică (RMN) sunt metode mai sensibile - permit diagnosticarea leziunilor osoase (osteopenie, infiltrare a măduvei osoase) în etapele incipiente care nu sunt disponibile pentru imagistică prin radiografie;
· Ecografia și RMN-ul ficatului și splinei pot dezvălui leziunile lor focale și pot determina volumul inițial de organe, necesar pentru monitorizarea ulterioară a eficacității terapiei de substituție enzimatică;
· Ecocardiografie Doppler - la pacienții splenectomizați;
esofagogastroduodenoscopie - în prezența unor plângeri adecvate sau semne de hipertensiune portală.
Diagnostic diferentiat
Diagnostic diferentiat
Boala Gaucher trebuie diferențiată de toate bolile care apar cu hepatosplenomegalie, citopenie, sângerare și leziuni osoase.
| Diagnostic | Motivarea diagnosticului diferențial | Sondaje | Criterii de excludere a diagnosticului |
| Hemoblastoză și limfoame | S-m hemoragic, dureri osoase, hepatosplenomegalie, |
2. mielogramă, |
|
| Anemii aplastice dobândite | S-m hemoragic, (+ / _) dureri osoase, pancitopenie |
1. Test general de sânge cu numărarea trombocitelor, reticulocitelor, 2. mielogramă, 3. test genetic de sânge molecular |
1. Absența unei scăderi a activității enzimei glucocerebrosidază și a unei creșteri a activității enzimei chitotriazidazei (în pete uscate de sânge prin spectrometrie de masă tandem sau fluorimetrie); 2. gena glucocerebrosidazei, localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31), nu a fost identificată; 3. Nu s-au detectat celule Gaucher la numărarea celulelor din mielogramă |
| Boală hepatică colestatică cronică, ciroză hepatică în rezultatul hepatitei cronice virale și non-virale | Hepatosplenomegalie, nivel crescut de transaminaze, bilirubină, s-m citopenic, s-m hemoragic, s-m dureros |
1. Test general de sânge cu numărarea trombocitelor, reticulocitelor, 2. mielogramă, 3. test genetic de sânge molecular 4. determinarea activității enzimelor glucocerebrosidază și chitriosidază 5. Test de sânge B / x 6. Ecografie, CT, RMN a organelor abdominale |
1. Absența unei scăderi a activității enzimei glucocerebrosidază și a unei creșteri a activității enzimei chitotriazidazei (în pete uscate de sânge prin spectrometrie de masă tandem sau fluorimetrie); 2. gena glucocerebrosidazei, localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31), nu a fost identificată; |
| Osteomielită cronică, tuberculoză osoasă | Ossalgia, limitarea mobilității membrelor |
2. mielogramă, 3. test genetic de sânge molecular 4. determinarea activității enzimelor glucocerebrosidază și chitriosidază 5. Test de sânge B / x |
1. Lipsa semnelor de citopenie (scăderea hemoglobinei, numărului de trombocite, leucopenie), 2. Absența unei scăderi a activității enzimei glucocerebrosidază și a unei creșteri a activității enzimei chitotriazidazei (în pete de sânge uscate prin spectrometrie de masă tandem sau fluorimetrie); 3. gena glucocerebrosidazei localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31) nu a fost identificată; 4. lipsa s-ma hemoragică, 5. Umflăturile caracteristice ale tibiei în formă de clavat sau balon („baloane Erlenmeyer”) nu sunt determinate de raze X. 5. Fără hepatosplenomegalie |
| Alte fermentopatii ereditare (boala Niemann-Pick |
Debutul precoce al dezvoltării bolii (3-5 luni), crește volumul abdomenului, dezvoltare psihomotorie întârziată, convulsii, alte simptome neurologice, dureri abdominale, sângerări, instabilitate emoțională |
1. Test general de sânge cu numărarea trombocitelor, reticulocitelor, 2. mielogramă, 3. test genetic de sânge molecular, (determinarea mutațiilor genei SMPD1, NPC1 și NPC2, gena glucocere brozidazei, localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31). 4. determinarea activității enzimelor glucocerebrosidază și chitriosidază, sfingomielinază 5. Test de sânge B / x 6. Ecografie, CT, RMN a organelor abdominale 7. Examinare cu raze X a țesutului osos (R, RMN, CT) 8. Examinarea de către un neurolog |
1. Absența unei scăderi a activității enzimei glucocerebrosidază și a unei creșteri a activității enzimei chitotriazidazei (în pete uscate de sânge prin spectrometrie de masă tandem sau fluorimetrie); |
| Histiocitoza | Ossalgia, limitarea mobilității membrelor, pancitopenie, s-m hemoragică, hepatosplenomegalie, pneumonie, tendință la infecții |
1. Test general de sânge cu numărarea trombocitelor, reticulocitelor, 2. mielogramă, imunofenotipare a măduvei osoase 3. test genetic de sânge molecular 4. determinarea activității enzimelor glucocerebrosidază și chitriosidază 5. Test de sânge B / x 6. Ecografie, CT, RMN a organelor abdominale 7. Examinare cu raze X a țesutului osos (R, RMN, CT) |
1. Absența unei scăderi a activității enzimei glucocerebrosidază și a unei creșteri a activității enzimei chitotriazidazei (în pete uscate de sânge prin spectrometrie de masă tandem sau fluorimetrie); 2. gena glucocerebrosidazei, localizată pe brațul lung al cromozomului 1 (regiunea 1q21q31), nu este detectată; 3 .. Radiografia nu determină umflarea caracteristică a clubului sau a balonului tibiei („baloane Erlenmeyer”). |
Tratament în străinătate
A urmat tratament în Coreea, Israel, Germania, SUA
Obțineți sfaturi despre turismul medical
Tratament
Preparate (ingrediente active) utilizate în tratament
| Azitromicină (azitromicină) |
| Alfakaltsidol (Alfakaltsidol) |
| Amfotericina B (Amfotericina B) |
| Aciclovir |
| Vancomicină (Vancomicină) |
| Voriconazol |
| Gentamicina |
| Diclofenac (Diclofenac) |
| Ibuprofen (Ibuprofen) |
| Imiglucerase |
| Imunoglobulină G (imunoglobulină G) |
| Iodixanol (Iodixanol) |
| Caspofungin |
| Clindamicina |
| Kolekaltsiferol (Kolekaltsiferol) |
| Lactuloza (Lactuloza) |
| Lornoxicam |
| Meropenem |
| Metronidazol (Metronidazol) |
| Micafungin |
| Complex ossein-hidroxiapatit |
| Paracetamol (Paracetamol) |
| Tramadol (Tramadol) |
| Fluconazol (Fluconazol) |
| Cefotaxime |
| Ceftazidime |
| Ceftriaxonă |
Tratament (ambulatoriu)
TRATAMENT LA NIVELUL AMBULATOR
Tacticile de tratament
Pacienții cu toate tipurile (I, II, III) de boală Gaucher primesc tratament ambulatoriu.
Tratament non-medicamentos:
· Mod - medical și de protecție în perioada s-ma citopenică, hemoragică, a complicațiilor osoase;
· Prevenirea leziunilor, reabilitarea focarelor cronice de infecție;
· Corecție psihologică - psihoterapie, adaptare psihologică.
Tratament medicamentos
Tratamentul modern al HD constă în prescrierea terapiei de substituție enzimatică pe tot parcursul vieții (ERT) cu glucocerebrosidază recombinantă, care ameliorează principalele manifestări clinice ale bolii, îmbunătățind calitatea vieții pacienților cu HD și fără a provoca reacții adverse pronunțate. . Fiecare pacient cu manifestări clinice de HD (HD de tip 1, HD de tip 3) trebuie să primească ERT. Doza de medicament trebuie selectată individual, în conformitate cu parametrii clinici și de laborator. În legătură cu dezvoltarea diagnosticului de laborator, la examinarea fraților (fraților și surorilor probandului) pot fi identificați copiii cu HD care nu au manifestări clinice. Acești pacienți au nevoie de supraveghere, dar tratamentul lor trebuie început numai atunci când apar simptomele bolii.
ERT își propune să furnizeze suficientă enzimă pentru a descompune depozitele de deșeuri. Astfel, terapia de substituție enzimatică funcționează prin adăugarea sau înlocuirea unei enzime lipsă sau defectă la pacienții cu boala Gaucher.

Lista medicamentelor esențiale
Imiglucerase
Tratamentul patogenetic al bolii Gaucher constă în prescrierea pe tot parcursul vieții a terapiei de substituție enzimatică cu glucocerebrosidază recombinantă. Doza inițială de imiglucerază per administrare în GD de tip I este de 30-40 UI / kg fără leziuni scheletice și 60 UI / kg în prezența leziunii osoase. La tipul III la copii, doza poate ajunge până la 100-120 unități / kg .
Medicamentul este administrat prin picurare intravenoasă la intervale de 1 la fiecare 2 săptămâni. (De 2 ori pe lună).
O scădere treptată a dozei cu 10-20 unități / kg este posibilă cu o dinamică pozitivă pronunțată după 1 an de tratament cu tip 1 BG fără deteriorarea oaselor și după 3-4 ani cu o deteriorare inițială a scheletului. Terapie de întreținere 15-60 unități / kg IV picurare timp de 3 ore la fiecare 2 săptămâni, pe viață.
Protocolul de terapie de substituție enzimatică imiglucerază

Lista medicamentelor suplimentare
Paracetamol
Lornaxicam
Diclofenac
Tramadol
Alfacalcidol
Flucanazol
Calciu Dz
Osteogenon
Aciclovir
Lactuloza
Cefotaxime
Ceftazidime
Ceftriaxonă
Azitromicina
Gentamicina
Iodixanol
Meropenem
Medicamente antiinflamatoare nesteroidiene:
Paracetamol - tablete 200 mg, 500 mg; lumânări. Adulți 500 mg de 3-4 ori pe zi timp de 3-7 zile. Copii în proporție de 60 mg / kg / zi în 3-4 doze, 3-7 zile;
Ibuprofen comprimate 200 mg, 400 mg; Copii - ibuprofen 30-40 mg / kg / zi,
Lornaxicam - 4 mg, 8 mg comprimate filmate. Adulți, 8 mg de 2 ori pe zi, pe cale orală, 2 săptămâni; liofilizat pentru prepararea unei soluții pentru administrare intravenoasă și intramusculară, 8 mg. Adulți, 8 mg de 2 ori pe zi, i / m, 10 zile;
· Diclofenac - soluție injectabilă 2,5% în fiole de 3 ml, tablete de 0,05 g, tablete retard de 0,025; 0,05 și 0,1 g; pastile pentru 0,025 g. Supozitoare rectale pentru 0,05 și 0,1 g. Gel, smântână, emulgel (1 g - 0,01 g de ortofen) în tuburi. Copii 2-3 mg / kg / zi, i.m., timp de 1-3-5 zile. Adulți de 7 mg de 2 ori pe zi, i.m., 1-3-5 zile.
Tramadol - soluție injectabilă 50 mg / ml, supozitoare rectale 0,1 g, picături -2,5 mg / capac, capsule 50 mg. În interior, doza inițială uzuală pentru adulți și copii cu vârsta peste 14 ani este de 50 mg (din nou, în absența efectului, după 30-60 de minute). Parenteral (i / v, i / m, s / c) - 50-100 mg, rectal - 100 mg (administrarea repetată de supozitoare este posibilă după 4-8 ore). Doza zilnică maximă este de 400 mg (în cazuri excepționale, poate fi crescută la 600 mg). Copii cu vârsta cuprinsă între 1 și 14 ani pe cale orală (picături) sau parenteral - o doză unică de 1-2 mg / kg, doza maximă zilnică este de 4-8 mg / kg.
Corectori ai metabolismului oaselor și cartilajelor:
Alfacalcidol 0,5mkg capsule. Doza zilnică pentru adulți variază de la 0,07 mcg la 20 mcg, pentru copii 0,01-0,08 mcg / kg. Doza zilnică pentru copii este de 0,01-0,08 mcg / kg.
· Calciu D3 - tablete masticabile care conțin (ingrediente active): carbonat de calciu - 1250 mg (corespunde 500 mg de calciu elementar); colecalciferol - 200 UI (unități internaționale). Adulți și copii cu vârsta peste 12 ani - 2 comprimate pe zi, în principal la mese.
Osteogenon - tablete de complex ossein-hidroxiapatit - 830mg; 2-4 file de 2 ori pe zi.
Algoritmul acțiunii în caz de situații urgente

Intervenție chirurgicală: Nu.
Alte tratamente:
· Reabilitare psihosocială: psihoterapie, adaptare psihologică, terapie de mediu;
· Adaptarea socială și îmbunătățirea calității vieții.
Indicații pentru consultarea de specialitate :
| Specialist | Indicaţie |
| traumatolog - ortoped |
Excluderea prezenței patologiei scheletice la un copil |
| Neuropatolog, psiho-neurolog | evaluarea stării neurologice, a stării neuropsihice, determinarea tipului de boală |
| kinetoterapeut |
determinarea metodelor de tratament fizioterapic |
| medic terapie exercițiu | selectarea unui program individual de exerciții de fizioterapie |
| genetician | confirmarea diagnosticului, genotiparea |
| Dacă este necesar, este posibilă consultarea cu alți specialiști, în funcție de cazul clinic. | |
Acțiuni preventive:
· Diagnosticul precoce al manifestărilor clinice ale bolii Gaucher pentru prevenirea complicațiilor;
· Consilierea genetică medicală pentru a explica riscul genetic.
· Prevenirea complicațiilor infecțioase pe fondul sindromului citopenic prelungit sunt, în unele cazuri, principalul motiv, în unele cazuri, chiar și decesul unui pacient.
· Îngrijirea cavității bucale: de 6-10 ori pe zi clătind cavitatea bucală cu soluții dezinfectante destinate tratamentului mucoasei bucale. Îngrijirea minuțioasă, dar blândă a dinților și a gingiilor; limitarea utilizării chiar a periuțelor moi de dinți; dați preferință dușului oral; cu trombocitopenie sau cu membrane mucoase vulnerabile, ar trebui exclusă utilizarea periuțelor de dinți, în schimb, este necesară curățarea suplimentară a gurii cu astringenți.
Când apar semne de stomatită, este necesar să se adauge la terapia de bază:
Fluconazol - doză estimată 4-5 mg / kg pe zi, capsule 50 mg, 100 mg, 150 mg, soluții perfuzabile 2 mg / ml, gel pentru tratament oral p, o IV,
· Aciclovir - doză estimată 250 mg / m2 x de 3 ori pe zi, tablete 200 mg, soluție injectabilă 250 mg, unguent pentru uz extern.
Dacă apar defecte ale mucoasei bucale: excludeți utilizarea periuțelor de dinți
2) odată cu dezvoltarea stomatitei necrotizante pe scară largă, este indicată terapia sistemică antifungică și antibacteriană:
· Cefotaxime, flacon de 1 g pentru prepararea soluției. Adulți 1-2g, de 2-3 ori pe zi, i / v, i / m, 7-10 zile. Copii 50-100 mg / kg greutate corporală / zi, de 2-4 ori pe zi, i / m, i / v, 7-10 zile;
Ceftazidime, flacon 250mg, 500mg, 1g, 2g pentru prepararea soluției. Adulți: 1-6 g / zi în 2 sau 3 doze intravenos sau intramuscular. Copii cu vârsta peste 2 luni: 30-100 mg / kg / zi în 2-3 doze, cu imunitate redusă - până la 150 mg / kg / zi (maxim 6 g / zi) în 3 doze. Pentru nou-născuți și sugari până la 2 luni: 25-60 mg / kg / zi în 2 doze divizate.
· Ceftriaxonă, flacon 500mg, 1g pentru prepararea soluției. Copii 50-80mg / kg / zi capac IV 1 oră 7-10 zile;
Iodixanol, soluție injectabilă, 100 mg / 2 ml și 500 mg / 2 ml. Adulții și copiii de la vârsta de 12 ani sunt prescriși intramuscular, intravenos (jet, în 2 minute sau prin picurare) la 5 mg / kg la fiecare 8 ore sau 7,5 mg / kg la fiecare 12 ore timp de 7-10 zile.
· Gentamicină, soluție injectabilă, fiole de 40 mg / ml. adulți 3-5 mg / kg (doza zilnică maximă) în 3-4 doze, 7-10 zile. Copiii mici sunt prescriși numai din motive de sănătate cu infecții severe. Doza zilnică maximă pentru copiii de toate vârstele este de 5 mg / kg.
Capsule de azitromicină 250, 500 mg. Copiii care cântăresc mai mult de 10 kg cu o rată de: în prima zi - 10 mg / kg greutate corporală; în următoarele 4 zile - 5 mg / kg. Este posibil un tratament de 3 zile; în acest caz, o doză unică este de 10 mg / kg. (Doza inițială 30 mg / kg greutate corporală). Adulților cu infecții ale tractului respirator superior și inferior, infecții ale pielii și țesuturilor moi li se prescriu 0,5 g în prima zi, apoi 0,25 g din a 2-a până la a 5-a zi sau 0,5 g pe zi în decurs de 3 zile (doza de curs de 1,5 g).
· Meropenem, pulbere pentru prepararea soluției pentru administrare intravenoasă de 0,5 și 1,0 g. Pentru copiii cu vârsta cuprinsă între 3 luni și 12 ani, doza recomandată este de 10-20 mg / kg la fiecare 8 ore, în funcție de tipul și severitatea infecției, sensibilitatea agentului patogen și starea pacientului. La copiii cu o greutate mai mare de 50 kg, trebuie utilizată doza pentru adulți.
3) Decontaminarea intestinelor se efectuează la alegerea spitalului, este posibil să se refuze decontaminarea. Decontaminarea (terapia preventivă) este recomandată pentru leziunile intestinale inițiale. Pentru decontaminarea selectivă a intestinului:
Ciprofloxacină în doză de 20 mg / kg pe zi, 100 mg într-un flacon, 250 mg, 500 mg în comprimate, picături pentru ochi, picături pentru urechi;
4) Este imperativ să se respecte igiena personală pentru toți cei care au grijă de bolnavi - părinți și vizitatori, spălare constantă a mâinilor.
Tacticile terapiei de substituțieși în conformitate cu Ordinul nr. 666 „La aprobarea Nomenclaturii, Regulile pentru achiziționarea, prelucrarea, depozitarea, vânzarea sângelui, precum și Regulile pentru depozitare, transfuzie de sânge, componentele sale și produse din sânge din 6 martie 2011, Anexă la Ordinul nr. 417 Ordin din data de 29.05.2015 an.
Monitorizarea pacientului:
· FZT pe viață;
Control dinamic: 1 an - 1 dată în 3 luni, apoi 1 dată în 6 luni:
· Adaptare socială;
· Observarea de către un genetician a familiei unui pacient cu HD.
Indicatori de eficacitate a tratamentului:
· Îmbunătățirea / stabilizarea parametrilor hematologici (ameliorarea sindromului citopenic, lipsa dependenței de transfuziile de sânge);
Restabilirea nivelului de glucocerebrosidază, scăderea indicelui chitotriosidazei;
· Eliminarea s-ma dureroasă;
· Restaurarea țesutului osos;
· Îmbunătățirea / stabilizarea funcției organelor extra-abdominale (inimă, plămâni, ochi);
· Reducerea frecvenței infecțiilor respiratorii;
· Scăderea ratei de progresie a bolii;
îmbunătățirea calității vieții pacientului (restabilirea dezvoltării mentale, spirituale, fizice).
Tratament (ambulanță)
MĂSURI DE DIAGNOSTIC REALIZATE LA ETAPA DE URGENȚĂ
Măsuri de diagnostic:
· Colectarea anamnezei;
· Examinare fizică;
· Determinarea patologiei cardiace (pulsoximetrie, tensiune arterială, ritm cardiac, ECG).
Tratament medicamentos
· Resuscitarea cardiopulmonară conform indicațiilor;
· Terapie sindrom-simptomatică conform indicațiilor;
· Terapia cu oxigen;
· Prevenirea aspirației;
Terapie antiinflamatoare analgezică
Tratament (spital)
TRATAMENT STAȚIONAR
Tacticile de tratament: urmăriți nivelul ambulatoriu.
Tratament medicamentos: urmăriți nivelul ambulatoriu.
Tratamentul medical se efectuează în conformitate cu Protocoalele clinice pentru complicațiile la jocuri de noroc.
Terapia medicamentoasă este îmbunătățită atunci când apar complicații pe fondul unui sindrom citopenic de lungă durată, stratificarea unei infecții virale / bacteriene și progresia bolii de bază. Cele mai grave complicații care pun viața în pericol sunt complicațiile infecțioase. Prezența febrei la un pacient cu neutropenie (neutrofile< 500/мкл) считается однократное повышение температуры тела >37,9 0 Cu o durată mai mare de o oră sau mai multe creșteri (de 3-4 ori pe zi) până la 38 0 C. Având în vedere riscul ridicat de apariție fatală a infecției, febra la un pacient cu neutropenie este considerată prezența de infecție, care dictează inițierea imediată a terapiei empirice cu antibiotice și efectuarea unui sondaj pentru a clarifica natura infecției. Au fost propuse multe regimuri antibacteriene inițiale, a căror eficacitate este în general identică.
Dispoziții generale:
· La alegerea unei combinații inițiale de antibiotice, este necesar să se ia în considerare: rezultatele studiilor bacteriologice repetate în această clinică la alți pacienți; durata neutropeniei actuale, istoricul infecțios al pacientului, cursurile anterioare de antibiotice și eficacitatea acestora
· Odată cu apariția febrei, toate celelalte date clinice: hipotensiune arterială, hemodinamică instabilă sunt o indicație pentru prescrierea imediată a unei combinații de antibiotice: carbopenemuri (meropenem (sau imipenem / cilastatină)) + aminoglicozid (amikacină) + vancomicină.
· CVC de lungă durată și febră după spălare și / sau nu doar febră, dar frisoane extraordinare ® Vancomicina este deja în combinația inițială;
· Clinica enterocolitei cu diaree: la combinația inițială - vancomicină per os 20 mg / kg pe zi. Poate numirea Metronidazolului (per os și / sau iv)
Stomatită severă cu modificări inflamatorii ale gingiilor ® penicilină, clindamicină în combinație cu beta-lactam sau Meropenem /
Erupții cutanate caracteristice și / sau prezență de drusen fungice în urină și / sau leziuni caracteristice în ficat și splină pe sonografie®
· Amfotericină B - liofilizat pentru prepararea soluției. Doza inițială este de 0,5 mg / kg în prima zi, a doua zi - doza terapeutică completă este de 1 mg / kg o dată pe zi. Când se utilizează amfotericina B, este necesar să se monitorizeze funcția rinichilor și să se efectueze un test biochimic de sânge (electroliți, creatinină). Este necesară corectarea constantă a potasiului la valorile normale. În timpul perfuziei de amfotericină B, precum și în decurs de aproximativ 3-4 ore după perfuzie, pot fi observate reacții la administrarea medicamentului sub formă de febră, frisoane uriașe, tahicardie, care sunt oprite de analgezice. În cazul afectării funcției renale, este necesar să se utilizeze voriconazol, Cancidas, forme lipidice ale amfotorecinei B.
Voriconazol - comprimat de 50 mg, liofilizat pentru prepararea soluției 200 mg / flacon. SD 4-6 mg / kg.
Caspofungin - liofilizat pentru prepararea soluției perfuzabile 50 mg
Mikofungin - liofilizat pentru prepararea soluției perfuzabile 50 mg
Schimbarea antibioticelor, luând în considerare sensibilitatea florei izolate. Eficacitatea terapiei antibiotice inițiale trebuie evaluată după 72 de ore, cu toate acestea, este necesară întotdeauna o examinare detaliată repetată a unui astfel de pacient la intervale de 8-12 ore, cu o evaluare a stabilității hemodinamicii și a gradului de intoxicație, a aspectului a noilor focare infecțioase. Terapia cu antibiotice continuă până la rezolvarea neutropeniei și rezolvarea completă a tuturor focarelor infecțioase.
Cu aplazie profundă, riscul de complicații septice, imunizare pasivă cu imunoglobuline G - 0,1-0,2 g / kg / zi capac IV.
Lista medicamentelor esențiale:
Imiglucerază 30-60 U / kg capac IV 3 ore
Lista medicamentelor suplimentare:
Paracetamol
Lornaxicam
Diclofenac
Tramadol
Alfacalcidol
Flucanazol
Calciu Dz
Osteogenon
Aciclovir
Lactuloza
Cefotaxime
Ceftazidime
Ceftriaxonă
Azitromicina
Gentamicina
Iodixanol
Meropenem
Imunoglobulina G
Amfotericina B
Voriconazol
Caspofungin
Micofungină
Vancomicină
Metronidazol
Clindamicina
Intervenție chirurgicală:
· Corectarea fracturilor osoase patologice, a contracturilor articulare.
Alte tratamente:
· Reabilitare fizică: fizioterapie, gimnastică de remediere, masaj;
· Reabilitare psihosocială: psihoterapie, adaptare psihologică, terapie de mediu.
Indicații pentru consultarea specialiștilor restrânși: urmăriți nivelul ambulatoriu.
Indicații pentru transferul la unitatea de terapie intensivă și la unitatea de terapie intensivă:
· Starea decompensată a pacientului;
· Generalizarea procesului cu dezvoltarea complicațiilor care necesită monitorizare intensivă și terapie;
· Perioada postoperatorie;
· Dezvoltarea de complicații pe fondul chimioterapiei intensive, care necesită tratament și monitorizare intensivă.
Indicatori de eficacitate a tratamentului:
· Refacerea dezvoltării mentale, spirituale, fizice;
· Restabilirea mobilității, a capacității de lucru;
· Eliminarea sindromului durerii în primii 2 ani de terapie;
· Prevenirea crizelor osoase;
· Prevenirea dezvoltării osteonecrozei și colapselor subcondrale;
· Îmbunătățirea indicelui densității minerale osoase;
· Creșterea densității minerale osoase pe parcursul a 3 ani de terapie;
· Realizarea ratelor normale de creștere în conformitate cu standardele populației în termen de 3 ani de la terapie;
· Atingerea vârstei normale de debut a pubertății.
· Normalizarea numărului de sânge în primii 3 ani de terapie;
· Reducerea hepatosplenomegaliei;
· Îmbunătățirea stării organelor extra-abdominale (inimă, plămâni, ochi).
Management ulterior:
Odată cu stabilizarea stării, refacerea parametrilor hematologici, ameliorarea durerii, intoxicație, afecțiuni hemoragice, copilul este externat pentru tratament ambulatoriu sub supravegherea unui pediatru, un hematolog la locul de reședință pentru a continua terapia de substituție enzimatică sub controlul testului . O monitorizare suplimentară a stării pacientului este de a urmări nivelul ambulatoriu.
Spitalizare
Indicații pentru spitalizarea planificată
Spitalizarea de rutină este indicată pentru a verifica diagnosticul și pentru a ajusta doza terapiei de substituție enzimatică.
Indicații pentru spitalizarea de urgență
· Sindrom citopenic;
· Sindromul durerii severe („criza osoasă”);
· Fractura patologică a oaselor scheletului;
· Insuficiență respiratorie.
informație
Surse și literatură
- Proces-verbal al ședințelor Comisiei mixte pentru calitatea serviciilor medicale ale Ministerului Sănătății și Dezvoltării Sociale din Republica Kazahstan, 2016
- 1) Dinte N. V. „Boala Gaucher: prevalență, semiotică, calitatea vieții și fundamentarea clinică și economică a terapiei de substituție enzimatică” rezumat al doctoratului. Moscova 2010 2) Lukina E.A. „Boala Gaucher: starea actuală a lucrurilor” Rossiyskie Meditsinskie Vesti 2008, Volumul XIII, nr. 2 p. 51-56. 3) Belogurova M.B. "Patogeneza, tabloul clinic, diagnosticul și tratamentul bolii Gaucher." Pediatrie și Chirurgie Pediatrică. Nr. 3 2010, pp. 43-48. 4) Aerts J. M., van Weely S., Broot R. și colab. Patogenia tulburărilor de stocare lizozomală așa cum este ilustrată de boala Gaucher // J. Inher. Metab. Dis. - 1993. - Vol. 16. Nr. 2. - P.288-291. 5) Beutler E., Grabowski G. A., Scriver C. R., și colab. Bazele metabolice și moleculare ale bolii moștenite // McGraw-Hill, New York, 2001. - P. 3635-3668. 6) de Frost M., vom Dahl S., Weverling G. J., și colab. Incidența crescută a cancerului la adulți Boala Gaucher în Europa de Vest // Celule sanguine Mol. Dis. - 2006. - Vol. 36.– P.53-58. 7) Taddei T.H., Kacena K.A., Yang M., și colab. Natura progresivă nerecunoscută a bolii Gaucher N370S și evaluarea riscului de cancer la 403 pacienți // Am. J. Hematol. - 2009. - Vol. 84. Nr. 4. - P.208-214. 8) Boala Niederau C. Gaucher. Bremen: UNI-MED; 2006,84 p. 9) Zimran A., Kay A., Beutler E. și colab. Boala Gaucher: caracteristici clinice, de laborator, radiologice și genetice la 53 de pacienți. Medicină 1992; 71: 337-53. 10) Weinreb N. J. Boala Gaucher de tip I la pacienții vârstnici. Gaucher Clin. Persp. 1999; 7 (2): 1-8. 11) Vorobiev A.I. (ed.) Farmacoterapie rațională a bolilor sistemului sanguin. M.: Litter, 2009, 563-6. 12) A.V. Davydov „Boli de stocare lizozomală: boala Gaucher” Jurnalul Medical Siberian, 2009, nr. 5. S.9-14. 13) Mikosch P., Reed M., Baker R. și colab. Modificări ale metabolismului osos la șapte pacienți cu boală Gaucher tratați consecutiv cu imiglucerază și miglustat // Calcif. Țesut Int. - 2008. - Vol. 83, nr. 1. - P.43-54. 14) vom Dahl S., Poll L., Di Rocco M. și colab. Recomandări bazate pe dovezi pentru monitorizarea bolilor osoase și a răspunsului la terapia de substituție enzimatică la pacienții cu Gaucher // Current Med. Cercetare și opinie. - 2006. –Vol. 22. Nr. 6. - P.1045-1064. 15) Wenstrup R. J., Roca-Espiau M., Weinreb N. J., și colab. Aspecte scheletice ale bolii Gaucher: o revizuire // Br. J. Radiol. 2002. Vol. 75. - P.2-12. 16) Cox TM, Schofield JP. Boala Gaucher: trăsături clinice și istorie naturală. Hematologia clinică a lui Bailliere. 1997; 10 (4): 657-689. 17) Boala Grabowski G. Gaucher: enzimologie, genetică și tratament. În: Harris H, Hirshchorn K, eds. Progrese în genetică umană. New York, NY: Plenum Press; 1993; 21: 377-441. 18) Vorobiev A.I. (ed.) Farmacoterapie rațională a bolilor sistemului sanguin. M.: Litter, 2009, 563-6. 19) Panoul de evaluare a tehnologiei NIH privind boala Gaucher, boala Gaucher: probleme actuale în diagnostic și tratament, JAMA. 1996; 275: 548-553. Panoul de evaluare a tehnologiei NIH privind boala Gaucher, boala Gaucher: probleme actuale în diagnostic și tratament, JAMA. 1996; 275: 548-553. 20) Grabowski G. A. Fenotip, diagnosticul și tratamentul bolii Gausher, s // Lancet. 2008. - Vol. 372. # 9645.-P. 1263-1271. 21) Abdilova G.K., Boranbaeva R.Z., Omarova K.O. și colab. Liniile directoare „Diagnosticul și tratamentul modern al bolii Gaucher la copiii din Kazahstan”, Almaty 2015 pp. 26-27. 22). Ghidul medicului pentru diagnosticul, tratamentul și urmărirea bolilor metabolice moștenite, ed. N. Blau, M. Duran, K.M. Gibson, C. Dionisi-Vici. 2014) 23) „Ghiduri clinice federale pentru furnizarea de îngrijiri medicale copiilor cu boala Gaucher” Moscova, 2015
informație
ABREVIERI UTILIZATE ÎN PROTOCOL:
ALT - alanina aminotransferaza
AST - aotocolaminotransferază aspartică
BG - boala Gaucher
RMN - Imagistica prin rezonanță magnetică
KLA - hemogramă completă
OAM - analiza generală a urinei
Ecografie - examen cu ultrasunete
ERT - terapie de substituție enzimatică
ECG - electrocardiogramă
EchoCG - ecocardiografie
LPN - boli de stocare lizozomală
SNC - sistemul nervos central
ADN - acid dezoxiribonucleic
HS - sindrom hemoragic
VSH - viteza de sedimentare a eritrocitelor
Tomografie computerizată CT
LISTA DEZVOLTĂTORILOR PROTOCOLULUI:
1) Boranbaeva Riza Zulkarnaevna - doctor în științe medicale, director al Centrului științific pentru pediatrie și chirurgie pediatrică.
2) Abdilova Gulnara Kaldenovna - candidat la științe medicale, director adjunct al Centrului de cercetare pentru pediatrie și chirurgie pediatrică, pediatrie.
3) Omarova Kulyan Omarovna - doctor în științe medicale, profesor, cercetător șef al Centrului științific pentru pediatrie și chirurgie pediatrică.
4) Manzhuova Lyazat Nurbapaevna - Candidat la științe medicale, șef al Departamentului de oncohematologie pentru copiii mai mari al Centrului științific pentru pediatrie și chirurgie pediatrică.
5) Satbayeva Elmira Maratovna - Candidat la științe medicale, RSE la REM „Universitatea Națională de Medicină Kazahă numită după SD Asfendiyarov”, șef al Departamentului de farmacologie.
INDICAREA FĂRĂ CONFLICT DE INTERES: absent.
REVIZORI:
1. Kurmanbekova Saule Kaspakovna - profesor al Departamentului de Internship și rezidențiat în pediatrie nr. 2 al Universității Naționale de Medicină din Kazahstan. S.D. Asfendiyarov.
INDICAREA CONDIȚIILOR PENTRU REVIZUIREA PROTOCOLULUI: revizuirea protocolului la 3 ani de la intrarea sa în vigoare și / sau când devin disponibile noi metode de diagnostic / tratament cu un nivel mai ridicat de dovezi.
Fișiere atașate
Atenţie!
- Automedicația poate provoca daune ireparabile sănătății dumneavoastră.
- Informațiile postate pe site-ul MedElement și în aplicațiile mobile „MedElement”, „Lekar Pro”, „Dariger Pro”, „Boli: Ghidul terapeutului” nu pot și nu ar trebui să înlocuiască o consultație personală cu un medic. Asigurați-vă că contactați un furnizor de asistență medicală dacă aveți afecțiuni sau simptome medicale care vă deranjează.
- Alegerea medicamentelor și dozarea acestora trebuie discutate cu un specialist. Numai un medic poate prescrie medicamentul necesar și dozajul acestuia, luând în considerare boala și starea corpului pacientului.
- Site-ul MedElement și aplicațiile mobile „MedElement”, „Lekar Pro”, „Dariger Pro”, „Boli: Ghidul terapeutului” sunt exclusiv informații și resurse de referință. Informațiile postate pe acest site nu trebuie utilizate pentru modificări neautorizate ale prescripțiilor medicului.
- Editorii MedElement nu sunt responsabili pentru orice daune aduse sănătății sau daunelor materiale rezultate din utilizarea acestui site.
