7.1. СТРУКТУРА, ВРЪЗКИ И ФУНКЦИИ НА МАЛЪЧНИЯ МОЗЪК
Малкият мозък (cerebellum) се намира под дубликат на твърдата обвивка на мозъка, известен като очертания на малкия мозък(tentorium cerebelli), който разделя черепната кухина на две неравномерни пространства – супратенториално и субтенториално. V субтенториално пространство,дъното на която е задната черепна ямка, в допълнение към малкия мозък, е мозъчният ствол. Обемът на малкия мозък е средно 162 cm 3. Теглото му варира между 136-169 g.
Малкият мозък се намира над моста и продълговатия мозък. Заедно с горното и долното мозъчно платно, той съставлява покрива на четвъртата камера на мозъка, дъното на която е така наречената ромбовидна ямка (виж глава 9). Над малкия мозък се намират тилните дялове на големия мозък, отделени от него от тенториума на малкия мозък.
В малкия мозък има две полукълба(hemispherum cerebelli). Между тях, в сагиталната равнина над IV вентрикул на мозъка, е разположена филогенетично най-древната част на малкия мозък - неговата червей(vermis cerebelli). Червените и малките полукълба са фрагментирани на лобули чрез дълбоки напречни жлебове.
Малкият мозък се състои от сиво и бяло вещество. Сивото вещество образува кората на малкия мозък и разположените в дълбочината му сдвоени ядра nuclei cerebelli (фиг. 7.1). Най-големите от тях са назъбени ядки(nucleus dentatus) - намира се в полукълба. В централната част на червея има тентови ядра(ядра
Ориз. 7.1.Церебеларни ядра.
1 - зъбно ядро; 2 - коркова сърцевина; 3 - сърцевината на палатката; 4 - сферично ядро.
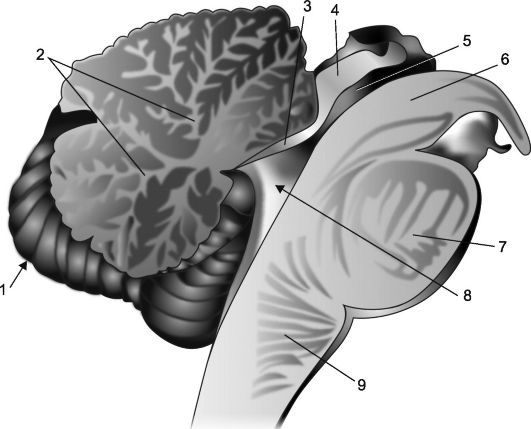
Ориз. 7.2.Сагитален разрез на малкия мозък и мозъчния ствол.
1 - малък мозък; 2 - "дърво на живота"; 3 - платно на предния мозък; 4 - плоча на четворката; 5 - акведукт на мозъка; 6 - крака на мозъка; 7 - мост; 8 - IV камера, нейният хороиден плексус и палатка; 9 - продълговатия мозък.
fastigii), между тях и зъбчатите ядра са сферичнаи коркови ядра(nuctei.globosus et emboliformis).
Поради факта, че кората покрива цялата повърхност на малкия мозък и прониква в дълбините на неговите бразди, на сагитален участък на малкия мозък, неговата тъкан има листен модел, чиито вени са образувани от бяло вещество (фиг. 7.2), което съставлява т.нар дървото на живота на малкия мозък (arbor vitae cerebelli). В основата на дървото на живота има клиновидна изрезка, която е горната част на кухината на IV камера; ръбовете на тази вдлъбнатина образуват палатката му. Покривът на палатката е мозъчният червей, а предната и задната му стени са тънки мозъчни пластини, известни като предна и задна мозъчни платна(vella medullare anterior et posterior).
Малко информация за мозъчна архитектоника,даващи основание за преценка на функцията на съставните му части. Имайте мозъчна кораИма два клетъчни слоя: вътрешният е гранулиран, състоящ се от малки зърнести клетки, а външният е молекулен. Между тях има редица големи крушовидни клетки, носещи името на чешкия учен И. Пуркине, който ги описва (Purkinje I., 1787-1869).
Импулсите навлизат в кората на малкия мозък през мъхести и пълзящи влакна, проникващи в нея от бялото вещество, които изграждат аферентните пътища на малкия мозък. Чрез мъхести влакна, импулси от гръбначния мозък
вестибуларните ядра и ядрата на моста се пренасят в клетките на гранулирания слой на кората. Аксоните на тези клетки, заедно с пълзящи влакна, преминаващи през гранулирания слой при транзит и пренасящи импулси от долните маслини към малкия мозък, достигат до повърхностния, молекулен слой на малкия мозък. Тук аксоните на клетките на гранулирания слой и пълзящите влакна се разделят в Т-образна форма, а в молекулярния слой техните разклонения поемат посока, надлъжна на повърхността на малкия мозък. Импулсите, достигнали до молекулярния слой на кората, след като са преминали през синаптичните контакти, попадат върху разклонените дендрити на клетките на Пуркине, разположени тук. След това те следват дендритите на клетките на Пуркине до телата им, разположени на границата на молекулярния и гранулирания слой. След това, по протежение на аксоните на същите клетки, пресичащи гранулирания слой, те проникват в дълбочината на бялото вещество. Аксоните на клетките на Пуркине завършват в ядрата на малкия мозък. Главно в зъбчатото ядро. Еферентните импулси, идващи от малкия мозък по аксоните на клетките, изграждащи ядрото му и участващи в образуването на малките дръжки, напускат малкия мозък.
Малкият мозък има три чифта крака:отдолу, средно и отгоре. Долният крак го свързва с продълговатия мозък, средният - с моста, горният - със средния мозък. Краката на мозъка изграждат пътищата, които пренасят импулси към и от малкия мозък.
Малкомозъчният червей осигурява стабилизиране на центъра на тежестта на тялото, неговото равновесие, стабилност, регулиране на тонуса на реципрочните мускулни групи, главно на врата и багажника, и възникването на физиологични мозъчни синергии, които стабилизират баланса на тялото.
За да поддържа успешно баланса на тялото, малкият мозък постоянно получава информация, преминаваща по спиноцеребеларните пътища от проприорецепторите на различни части на тялото, както и от вестибуларните ядра, долните маслини, ретикуларната формация и други формации, участващи в контрола на положение на частите на тялото в пространството. Повечето от аферентните пътища, водещи към малкия мозък, преминават през долната мозъчна педикула, някои от тях са разположени в горната мозъчна педикула.
Импулси на проприоцептивна чувствителност, отивайки към малкия мозък, подобно на други сензорни импулси, следвайки дендритите на първите сензорни неврони, достигат телата им, разположени в гръбначните възли. Впоследствие импулси, отиващи към малкия мозък по аксоните на същите неврони, се насочват към телата на втори неврони, които се намират във вътрешните части на основата на задните рога, образувайки т.нар. Стълбовете на Кларк. Аксоните им попадат в страничните участъци на страничните струни на гръбначния мозък, където образуват спиноцеребеларни пътища, в този случай част от аксоните попадат в страничната колона от същата страна и се образуват там задния спиноцеребеларен тракт на Flexig (tractus spinocerebellaris posterior). Друга част от аксоните на клетките на задните рога преминава от другата страна на гръбначния мозък и навлиза в противоположния страничен мозък, образувайки в него преден спиноцеребеларен тракт на Govers (tractus spinocerebellaris anterior). Спиноцеребеларните пътища, увеличавайки обема си на нивото на всеки гръбначен сегмент, се издигат до продълговатия мозък.
В продълговатия мозък задният спиноцеребеларен път се отклонява в странична посока и, преминавайки през долната мозъчна педикула, прониква в малкия мозък. Предният спиномозъчен път минава през продълговатия мозък, моста на мозъка, и достига до средния мозък, на нивото на който прави второто си пресичане в предния мозъчен велум и преминава в малкия мозък през горната мозъчна дръжка.
Така от двата гръбначно-мозъчни тракта единият никога не се пресича (непреминава пътя на Флексиг), а другият минава на противоположната страна два пъти (два пъти пресечен от Гоувърс). В резултат и двете провеждат импулси от всяка половина на тялото, главно към хомолатералната половина на малкия мозък.
В допълнение към спиноцеребеларните пътища на Fleksig, импулсите към малкия мозък преминават през долния мозъчен педикул по протежение на вестибулоцеребеларния тракт (tractus vestibulocerebellaris), започващ главно в горното вестибуларно ядро на анкилозиращ спондилит и по протежение на оливомоцеребеларния тракт (tractus olivocerebellaris), идващ от долната маслина. Част от аксоните на клетките на тънките и клиновидни ядра, не участва в образуването на булботаламичния тракт, под формата на външни дъговидни влакна (fiber arcuatae externae) също навлиза в малкия мозък през долната дръжка на малкия мозък.
Чрез средните си крака малкият мозък получава импулси от кората на главния мозък. Тези импулси преминават кортикално-церебелопонтинни пътища, състоящи се от два неврона. Телата на първите неврони са разположени в кората на главния мозък, главно в кората на задните части на челните лобове. Аксоните им преминават като част от лъчистата корона, предния крак на вътрешната капсула и завършват в ядрата на моста. Аксони на клетки на втори неврони, чиито тела са разположени в собствените си ядра на моста, отидете на противоположната му страна и съставете, след пресечната точка, средната мозъчна ножка,
завършващ в противоположното полукълбо на малкия мозък.
Част от импулсите, възникнали в кората на главния мозък, достигат до противоположното полукълбо на малкия мозък, като носят информация не за произведеното, а само за планираното активно движение. След като получи такава информация, малкият мозък незабавно изпраща импулси, които коригират волевите движения, главно, чрез гасене на инерцията и най-рационалното регулиране на реципрочния мускулен тонус - мускулни агонисти и антагонисти. В резултат на това един вид ейметрия,правене на волевите движения ясни, усъвършенствани, лишени от неподходящи компоненти.
Пътищата, които напускат малкия мозък, са съставени от аксоните на клетките, чиито тела образуват нейните ядра. Повечето еферентни пътища, включително пътища от зъбчатите ядра, напускат малкия мозък през горния му крак. На нивото на долните туберкули на четворката се пресича еферентният мозъчен тракт (пресечна точка на горните мозъчни крака на Вернекинг). След преминаване на всеки един от тях достига до червените ядра на противоположната страна на средния мозък. В червените ядра мозъчните импулси преминават към следващия неврон и след това се движат по аксоните на клетките, чиито тела са вградени в червените ядра. Тези аксони се образуват в червено-ядрено-гръбначно-мозъчни пътища (tracti rubro spinalis), пътищата на Монаков, които скоро след това изходите от червените ядки се кръстосват (кръст на гуми или кръст на пъстърва), след което те се спускат в гръбначния мозък. В гръбначния мозък червено-ядрените гръбначни пътища са разположени в страничните мозъци; съставните им влакна завършват при клетките на предните рога на гръбначния мозък.
Целият еферентен път от малкия мозък до клетките на предните рога на гръбначния мозък може да се нарече мозъчен-червен-ядрено-гръбначен (tractus cerebello-rubrospinalis). Той пресича два пъти (пресичане на горните мозъчни дръжки и пресичане на оперкулума) и в крайна сметка свързва всяко полукълбо на малкия мозък с периферни моторни неврони, разположени в предните рога на хомолатералната половина на гръбначния мозък.
От ядрата на мозъчния червей еферентните пътища преминават главно през долната мозъчна педикула до ретикуларната формация на мозъчния ствол и вестибуларните ядра. Оттук по ретикулоспиналните и вестибулоспиналните пътища, минаващи по предните струни на гръбначния мозък, те достигат и до клетките на предните рога. Част от импулсите, идващи от малкия мозък, преминавайки през вестибуларните ядра, навлизат в медиалния надлъжен сноп, достигат до ядрата III, IV и VI на черепните нерви, които осигуряват движението на очните ябълки, и засяга тяхната функция.
В обобщение трябва да се подчертае следното:
1. Всяка половина на малкия мозък получава импулси основно а) от хомолатералната половина на тялото, б) от противоположното полукълбо на мозъка, което има кортико-гръбначни връзки със същата половина на тялото.
(2) От всяка половина на малкия мозък се насочват еферентни импулси към клетките на предните рога на хомолатералната половина на гръбначния мозък и към ядрата на черепните нерви, които осигуряват движението на очните ябълки.
Това естество на мозъчните връзки дава възможност да се разбере защо, когато е засегната едната половина на малкия мозък, мозъчните разстройства възникват главно в същия, т.е. хомолатерална, половината от тялото. Това е особено изразено, когато са засегнати полукълба на малкия мозък.
7.2. ИЗСЛЕДВАНЕ НА ФУНКЦИИТЕ НА МОЗЪЧКАТА
И КЛИНИЧНИ ПРОЯВИ НА НЕГОВИТЕ ПОРАЖЕНИЯ
При увреждане на малкия мозък са характерни нарушения на статиката и координацията на движенията, мускулна хипотония и нистагъм.
Лезия на малкия мозък преди всичко неговият червей,води до нарушения на статиката - способността да се поддържа стабилно положение на центъра на тежестта на човешкото тяло, баланс, стабилност. Когато тази функция е нарушена, статична атаксия (от гръцки атаксия - разстройство, нестабилност). Отбелязва се нестабилността на пациента. Затова в изправено положение той разтваря краката си широко, балансира с ръце. Особено ясно се открива статична атаксия с изкуствено намаляване на областта на опората, по-специално в позата на Ромберг. Пациентът е поканен да се изправи, като движи здраво краката си и леко повдига главата си. При наличие на мозъчни нарушения се отбелязва нестабилността на пациента в това положение, тялото му се люлее, понякога се „дърпа“ в определена посока и ако пациентът не се поддържа, той може да падне. В случай на увреждане на мозъчния червей, пациентът обикновено се люлее от едната към другата страна и често пада назад. При патологията на полукълбото на малкия мозък има тенденция към падане главно към патологичния фокус. Ако статичното разстройство е умерено изразено, по-лесно се идентифицира в т.нар сложенили чувствителна поза на Ромберг. Пациентът е помолен да постави краката си в една линия, така че пръстът на единия крак да лежи върху петата на другия. Оценката за стабилност е същата като в обичайната позиция на Ромберг.
Обикновено, когато човек е изправен, мускулите на краката му са напрегнати. (реакция на подкрепа), със заплахата да падне настрани, кракът му от тази страна се движи в същата посока, а другият крак се отделя от пода (реакция на скок). При увреждане на малкия мозък (главно на червея) реакциите на пациента са нарушени
подкрепа и скок. Нарушаването на опорната реакция се проявява чрез нестабилност на пациента в изправено положение, особено в позицията на Ромберг. Нарушаването на реакцията на скока води до факта, че ако лекарят, застанал зад пациента и го осигурява, избута пациента в една или друга посока, тогава пациентът пада с лек тласък (симптом на натискане).
При увреждане на малкия мозък походката на пациента обикновено се променя поради развитието статолокомоторна атаксия. Малкомозъчна походка в много отношения наподобява походката на пиян човек, поради което понякога се нарича "походка на пиян". Поради нестабилност пациентът ходи несигурно, разтваряйки широко крака, докато се „хвърля“ от едната страна на другата. А когато е увредено полукълбото на малкия мозък, то се отклонява при ходене от дадена посока към патологичния фокус. Нестабилността е особено изразена при завой. Ако атаксията е силно изразена, тогава пациентите напълно губят способността си да контролират тялото си и могат не само да стоят и да ходят, но дори и да седят.
Преобладаващото увреждане на полукълба на малкия мозък води до нарушение на неговите антиинерционни ефекти, по-специално до появата на кинетична атаксия. Проявява се с неудобството на движенията и е особено изразено при движения, които изискват прецизност. За откриване на кинетична атаксия се правят тестове за координация на движенията. Някои от тях са описани по-долу.
Тест за диадохокинеза (от гръцки diadochos - последователност). Пациентът се приканва да затвори очи, да протегне ръцете си напред и бързо, ритмично да супинира и да проникне в ръцете. В случай на увреждане на полукълбото на малкия мозък, движенията на ръката от страната на патологичния процес се оказват по-размахващи (последствие от дисметрия, по-точно хиперметрия), в резултат на това ръката започва да изостава. Това показва наличието на адиадохокинеза.
Тест с пръст. Пациент със затворени очи трябва да изтегли ръката си и след това бавно, с показалеца, да докосне върха на носа. В случай на мозъчна патология ръката отстрани на патологичния фокус прави прекомерно движение по обем (хиперметрия),в резултат на което пациентът пропуска. Тестът пръст-нос разкрива характеристика на мозъчната патология церебеларен (умишлен) тремор, чиято амплитуда се увеличава с приближаването на пръста към целта. Този тест ви позволява да идентифицирате така наречената брадителекинезия. (симптом на юзда):недалеч от целта, движението на пръста се забавя, понякога дори спира и след това се възобновява отново.
Тест пръст-пръст. Пациент със затворени очи се приканва да разпери ръцете си широко и след това да приближи показалците, опитвайки се да вкара пръста в пръста, докато, както при теста с пръст, се разкриват умишлен тремор и симптом на юзда.
Тест за калканеално коляно (фиг. 7.3). Пациентът, лежащ по гръб със затворени очи, е помолен да вдигне високо единия крак и след това да удари с петата си коляното на другия крак. При мозъчна патология пациентът не може или му е трудно да вкара петата си в коляното на другия крак, особено при извършване на тест с крака хомолатерално спрямо засегнатото полукълбо на малкия мозък. Ако все пак петата достигне коляното, тогава се предлага да я държите, леко докосвайки предната повърхност на подбедрицата, надолу до глезенната става, докато в случай на мозъчна патология петата се плъзга от долния крак. времето в едната или другата посока.

Ориз. 7.3.Тест за калканеално коляно.
Примерен тест: Пациентът се приканва да удари с показалеца си няколко пъти гумения връх на чука, който е в ръката на изследващия. В случай на мозъчна патология в ръката на пациента от страната на засегнатото полукълбо на малкия мозък, има несъответствие поради дисметрия.
Симптом на Том-Джументи: Ако пациентът вдигне предмет, например чаша, той прекомерно разтваря пръстите си.
Церебеларен нистагъм. Потрепването на очните ябълки при гледане встрани (хоризонтален нистагъм) се счита за последица от преднамерен тремор на очните ябълки (виж глава 30).
Разстройство на говора: Речта губи своята плавност, става експлозивна, фрагментирана, напява се като церебеларна дизартрия (виж глава 25).
Промяна на почерка: Поради нарушение в координацията на движенията на ръцете, почеркът става неравномерен, буквите са деформирани, прекомерно големи (мегалография).
Пронаториален феномен: Пациентът е помолен да държи ръцете си изпънати в супинация, докато скоро настъпва спонтанна пронация от страната на засегнатото полукълбо на малкия мозък.
Симптом на Хоф-Шилдер: Ако пациентът държи ръцете си изпънати напред, тогава от страната на засегнатото полукълбо, ръката скоро се изтегля навън.
Феномен на имитация. Пациент със затворени очи трябва бързо да даде на ръката позиция, подобна на тази, която изпитващият преди това е дал на другата си ръка. Когато мозъчното полукълбо е увредено, хомолатералната ръка прави движение с прекомерна амплитуда.
Феноменът на Дойников. Феномен на пръстите. Седящият пациент е поканен да постави супинирани ръце с раздалечени пръсти върху бедрата си и да затвори очи. В случай на лезия на малкия мозък от страната на патологичното огнище, скоро настъпва спонтанна флексия на пръстите и пронация на ръката и предмишницата.
Симптом на Стюарт-Холмс. Изследователят моли пациента, седнал на стола, да огъне супинираните предмишници и в същото време, хващайки ръцете си за китките, му оказва съпротива. Ако в същото време внезапно освободите ръцете на пациента, тогава ръката от засегнатата страна, огъваща се по инерция, ще го удари със сила в гърдите.
Мускулна хипотония. Поражението на червата на малкия мозък обикновено води до дифузна мускулна хипотония. С поражението на полукълбото на малкия мозък, пасивните движения разкриват намаляване на мускулния тонус от страна на патологичния процес. Мускулната хипотония води до възможност за хиперекстензия на предмишницата и подбедрицата (симптом на Олшански) с пасивни движения, към външния вид симптоми на висяща ръка или крак с пасивното им разклащане.
Патологични асинергии на малкия мозък. Нарушенията на физиологичните синергии по време на сложни двигателни актове се разкриват по-специално по време на следните тестове (фиг. 7.4).
1. Асинергия според Бабински в изправено положение.Ако пациент, който стои с изместени крака, се опитва да се наведе назад, хвърляйки главата си назад, тогава нормално в този случай се получава флексия на коленните стави. При мозъчна патология поради асинергия това приятелско движение отсъства и пациентът, губейки равновесие, пада назад.
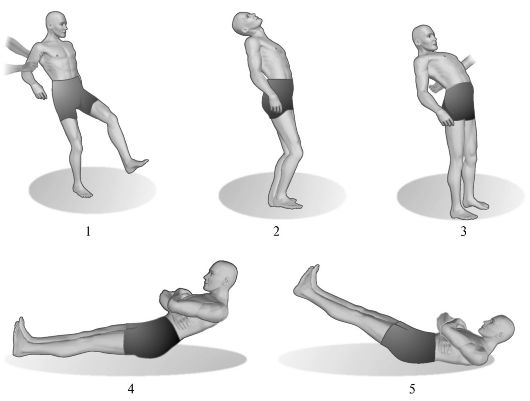
Ориз. 7.4.Церебеларна асинергия.
1 - походка на пациент с тежка церебеларна атаксия; 2 - наклонът назад на тялото е нормален; 3 - с увреждане на малкия мозък, пациентът, наведен назад, не може да поддържа равновесие; 4 - извършване на тест за мозъчна асинергия по Бабински от здрав човек; 5 - извършване на същия тест при пациенти с мозъчни лезии.
2. Асинергия според Бабински в легнало положение.Пациентът, лежащ на твърда равнина с изпънати крака, разперени до ширината на раменния пояс, се приканва да кръстоса ръце на гърдите си и след това да седне. При наличие на мозъчна патология поради липса на приятелско свиване на глутеалните мускули (проява на асинергия), пациентът не може да фиксира краката и таза върху опорната зона, в резултат на това краката се издигат и той не може да седне. Значението на този симптом не трябва да се надценява при пациенти в напреднала възраст, при хора с отпусната или затлъстяла коремна стена.
Обобщавайки горното, трябва да се подчертае разнообразието и важността на функциите, изпълнявани от малкия мозък. Като част от сложен регулаторен механизъм за обратна връзка, малкият мозък действа като фокусна точка за балансиране на тялото и поддържане на мускулния тонус. Както отбелязва П. Дуус (1995), малкият мозък осигурява способността за извършване на дискретни и прецизни движения,авторът основателно смята, че малкият мозък работи като компютър, проследявайки и координирайки сензорната информация на входа и симулирайки двигателни сигнали на изхода.
7.3. МУЛТИСИСТЕМНИ ДЕГЕНЕРАЦИИ
С признаци на мозъчна патология
Мултисистемните дегенерации са група невродегенеративни заболявания, общата черта на които е мултифокалният характер на лезията с участието на различни функционални и невротрансмитерни системи на мозъка в патологичния процес и следователно полисистемният характер на клиничните прояви.
7.3.1. Церебеларна атаксия
Спиноцеребеларните атаксии включват прогресиращи наследствени дегенеративни заболявания, при които се засягат основно структурите на малкия мозък, мозъчния ствол и пътищата на гръбначния мозък, които са свързани основно с екстрапирамидната система.
7.3.1.1. Наследствена атаксия на Фридрейх
Наследствено заболяване, описано през 1861 г. от немския невропатолог Н. Friedreich (Friedreich N., 1825-1882). Наследява се по автозомно рецесивен начин или (по-рядко) по автозомно доминантен начин с непълна пенетрантност и променлива генна експресия. Възможни са и спорадични случаи на заболяването.
Патогенезазаболяване не е посочено. По-специално, няма представа за първичния биохимичен дефект, който представлява неговата основа.
Патоморфология.Патологичните изследвания разкриват изразено изтъняване на гръбначния мозък поради атрофични процеси в задния и страничния му мозък. По правило страдат клиновидните (Бурдах) и нежните (Гол) пътища и спиноцеребеларните пътища на Говерс и Флексиг, както и кръстосаният пирамидален път, съдържащ
много влакна, свързани с екстрапирамидната система. Дегенеративните процеси се изразяват и в малкия мозък, в неговото бяло вещество и в ядрения апарат.
Клинични проявления. Заболяването се проявява при деца или млади хора под 25-годишна възраст. С.Н. Давиденков (1880-1961) отбелязва, че по-често клиничните признаци на заболяването се срещат при деца на възраст 6-10 години. Първият признак на заболяване обикновено е атаксия. Пациентите изпитват несигурност, залитане при ходене, промени в походката (при ходене краката са широко раздалечени). Походката при болестта на Фридрейх може да се нарече табетично-мозъчна, тъй като нейните промени са причинени от комбинация от чувствителна и церебеларна атаксия, както и обикновено изразено намаляване на мускулния тонус. Характерни са и нарушения на статиката, дискоординация в ръцете, умишлен тремор, дизартрия. Възможен е нистагъм, загуба на слуха, елементи на речта на пеене, признаци на пирамидална недостатъчност (хиперрефлексия на сухожилията, патологични рефлекси на краката, понякога леко повишаване на мускулния тонус), наложително желание за уриниране, намалена сексуална потентност. Понякога се появява атетоидна хиперкинеза.
Ранното разстройство на дълбоката чувствителност води до прогресивно намаляване на сухожилните рефлекси: първо на краката, а след това на ръцете. С течение на времето се образува мускулна хипотрофия на дисталните крака. Характерно е наличието на аномалии в развитието на скелета. На първо място, това се проявява чрез присъствието Краката на Фридрайх: стъпалото е скъсено, "кухо", с много висок свод. Основните фаланги на пръстите й са разгънати, останалите са огънати (фиг. 7.5). Възможна деформация на гръбначния стълб, гръдния кош. Често има прояви на кардиопатия. Заболяването прогресира бавно, но постоянно води до инвалидизация на пациентите, които в крайна сметка стават приковани на легло.
Лечение. Патогенетично лечение не е разработено. Предписвайте лекарства, които подобряват метаболизма в структурите на нервната система, укрепващи средства. При тежка деформация на краката са показани ортопедични обувки.
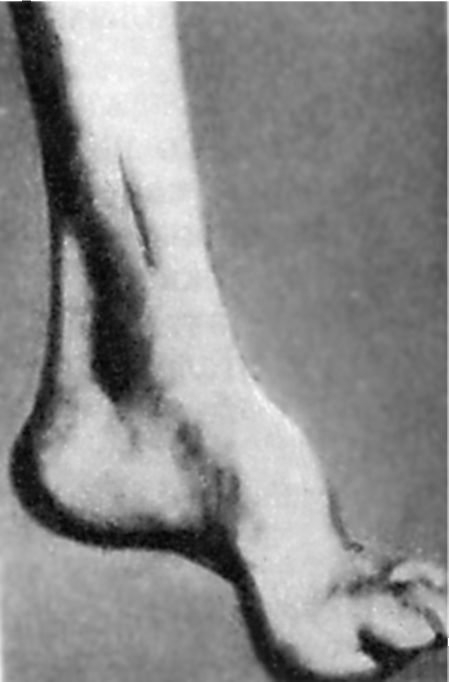
Ориз. 7.5.Кракът на Фридрейх.
7.3.1.2. Наследствена церебеларна атаксия (болест на Пиер Мари)
Това е хронично прогресиращо наследствено заболяване, проявяващо се на възраст 30-45 години, с бавно нарастващи мозъчни нарушения в съчетание с признаци на пирамидална недостатъчност, като са характерни статична и динамична мозъчна атаксия, интенционен тремор, скандирана реч, сухожилна хиперрефлексия. Възможни клонуси, патологични пирамидални рефлекси, страбизъм, намалено зрение, стесняване на зрителните полета поради първична атрофия на зрителните нерви и пигментна дегенерация на ретината. Ходът на заболяването е бавно прогресиращ. Наблюдава се намаляване на размера на малкия мозък, клетъчна дегенерация
Пуркине, долни маслини, спиноцеребеларни пътища. Наследява се по автозомно доминантен начин. Заболяването е описано през 1893 г. от френския невропатолог Р. Мари (1853-1940).
В момента няма единодушие в разбирането на термина "болест на Пиер Мари", а въпросът за възможността за отделянето му в самостоятелна нозологична форма е спорен.
Не е разработено лечение. Обикновено се използват метаболитно активни и възстановяващи, както и симптоматични средства.
7.3.2. Оливопонтоцеребеларна дистрофия (болест на Дежерин-Том)
Това е група хронични прогресивни наследствени заболявания, при които дистрофичните изменения се развиват главно в малкия мозък, долните маслини, в собствените ядра на моста и в свързаните с тях мозъчни структури.
С развитието на заболяването в млада възраст около половината от случаите се унаследяват по доминантен или рецесивен начин, останалите са спорадични. При спорадични случаи на заболяването са по-чести прояви на акинетично-ригиден синдром и прогресираща автономна недостатъчност. Средната възраст на пациента с проява на наследствена форма на заболяването във фенотипа е 28 години, със спорадична - 49 години, средната продължителност на живота е съответно 14,9 и 6,3 години. При спорадична форма, освен атрофия на маслините, моста и малкия мозък, по-често се откриват лезии на страничните струни на гръбначния мозък, черната субстанция и стриатума, синкаво петно в ромбовидната ямка на четвъртата камера на мозъка .
Характерни са симптомите на нарастващия церебеларен синдром. Възможни са нарушения на чувствителността, елементи на булбарни и акинетично-ригидни синдроми, хиперкинези, по-специално миоритмии на езика и мекото небце, офталмопареза, намалена зрителна острота, интелектуални нарушения. Заболяването е описано през 1900 г. от френските невропатолози Ж. Дежерин и А. Томас.
Заболяването често дебютира с нарушения в ходенето – възможни са нестабилност, дискоординация, неочаквани падания. Тези нарушения могат да бъдат единствената проява на заболяването в рамките на 1-2 години. В бъдеще възникват и растат нарушения на координацията в ръцете: манипулациите с малки предмети са трудни, ръкописът е нарушен, възниква умишлен тремор. Речта става прекъсваща, замъглена, с назален оттенък и ритъм на дишане, който не съответства на структурата на речта (болният говори, сякаш го удушават). На този етап на заболяването се присъединяват прояви на прогресираща автономна недостатъчност, появяват се признаци на акинетично-ригиден синдром. Понякога доминиращите симптоми за пациента са дисфагия, пристъпи на нощно задушаване. Развиват се във връзка със смесена пареза на булбарната мускулатура и могат да бъдат животозастрашаващи.
През 1970 г. немските невропатолози B.W. Konigsmark и L.P. Уайнер изтъкна 5 основни видаоливопонтоцеребеларна дистрофия, различаваща се или по клинични и морфологични прояви, или по вида на унаследяване.
аз тип (тип Менцел). На възраст 14-70 (по-често 30-40) години се проявява атаксия, дизартрия, дисфония, мускулна хипотония, в късния стадий - силен тремор на главата, тялото, ръцете, мускулите, признаци на акинетично- ригиден синдром. Възможни патологични пирамидални признаци, пареза на погледа, външна и вътрешна офталмоплегия, нарушения на чувствителността, деменция. Наследява се по автозомно доминантен начин. Обособена е като самостоятелна форма през 1891 г. от П. Менцел.
II тип (тип Фиклер-Уинклер) ... На 20-80 години се проявява атаксия, понижен мускулен тонус и сухожилни рефлекси. Наследява се по автозомно рецесивен начин. Възможни са спорадични случаи.
III тип с дегенерация на ретината. Проявява се в детска или млада (до 35 години) атаксия, тремор на главата и крайниците, дизартрия, признаци на пирамидална недостатъчност, прогресивно намаляване на зрението с изход до слепота; възможен нистагъм, офталмоплегия, понякога дисоциирани сензорни нарушения. Наследява се по автозомно доминантен начин.
IV тип (тип Jester-Highmaker). На възраст 17-30 години той дебютира с церебеларна атаксия или признаци на долна спастична парапареза, и в двата случая, вече в ранния стадий на заболяването, се формира комбинация от тези прояви, към които се появяват елементи на булбарен синдром, пареза на лицевите мускули и впоследствие се добавят нарушения на дълбоката чувствителност. Доминантно наследено.
V тип. Проявява се на възраст 7-45 години атаксия, дизартрия, признаци на акинетично-ригиден синдром и други екстрапирамидни нарушения, прогресираща офталмоплегия и деменция. Доминантно наследено.
7.3.3. Оливоруброцеребеларна дегенерация (синдром на Lejeune-Lermitte, болест на Lermitte)
Заболяването се характеризира с прогресивна атрофия на малкия мозък, главно на неговия кортекс, назъбени ядра и горни мозъчни дръжки, долни маслини и червени ядра. Проявява се предимно със статична и динамична атаксия; в бъдеще са възможни други признаци на церебеларен синдром и увреждане на мозъчния ствол. Заболяването е описано от френските невропатолози J. Lhermitte (Lhermitte J.J., 1877-1959) и J. Lezhon (Lejonne J., роден през 1894 г.).
7.3.4. Мултисистемна атрофия
През последните десетилетия спорадичното, прогресивно невродегенеративно заболяване, наречено мултисистемна атрофия, беше идентифицирано като независима форма. Характеризира се с комбинирана лезия на базалните ганглии, малкия мозък, мозъчния ствол, гръбначния мозък. Основните клинични прояви: паркинсонизъм, церебеларна атаксия, признаци на пирамидална и автономна недостатъчност (Levin O.S., 2002). В зависимост от преобладаването на определени характеристики на клиничната картина се разграничават три вида мултисистемна атрофия.
1) оливопонтоцеребеларен тип, характеризиращ се с преобладаване на признаци на мозъчна атака;
2) стрионигрален тип, при който доминират признаците на паркинсонизъм;
3) Синдром на Shai-Drager, характеризиращ се с преобладаване в клиничната картина на признаци на прогресираща автономна недостатъчност със симптоми на ортостатична артериална хипотония.
В основата на мултисистемната атрофия е селективната дегенерация на определени области от преобладаващо сивото вещество на мозъка с увреждане на невроните и глиалните елементи. Причините за дегенеративните прояви в мозъчната тъкан остават неизвестни и днес. Проявите на мултисистемна атрофия от оливопонтоцеребеларния тип са свързани с увреждане на клетките на Пуркине в кората на малкия мозък, както и неврони на долните маслини, понтоцеребеларните ядра, демиелинизация и дегенерация, главно на понтоцеребеларните пътища.
Церебеларните нарушения обикновено са статична и динамична атаксия с нарушено двигателно движение. Характеризира се с нестабилност в позицията на Ромберг, атаксия при ходене, дисметрия, адиадохокинеза, преднамерен тремор, може да има нистагъм (хоризонтален вертикален, биещ надолу), периодични и бавни проследяващи движения на погледа, нарушена конвергенция на очите, скандирана реч.
Мултисистемната атрофия обикновено се появява в зряла възраст и прогресира бързо. Диагнозата се основава на клинични данни и се характеризира с комбинация от признаци на паркинсонизъм, мозъчна недостатъчност и вегетативни нарушения. Лечението на заболяването не е разработено. Продължителността на заболяването - в рамките на 10 години, завършва със смърт.
7.4. ДРУГИ ЗАБОЛЯВАНИЯ, СВЪРЗАНИ СЪС СИМПТОМИ НА МОЗЪЧНО ЗАБОЛЯВАНЕ
Ако пациентът показва признаци на мозъчна лезия, тогава в повечето случаи преди всичко трябва да помислиш за възможносттамозъчни тумори(астроцитом, ангиобластом, медулобластома, метастатични тумори) или множествена склероза. В мозъчни туморипоявяват се ранни признаци на вътречерепна хипертония. При множествена склероза обикновено е възможно да се идентифицират, в допълнение към патологията на малкия мозък, клинични прояви на увреждане на други структури на централната нервна система, предимно зрителната и пирамидната системи. В класическата неврология характеристиката на множествена склерозаТриада на Шарко: нистагъм, преднамерен тремор и скандирана реч и Синдром на Ноне:нарушение на координацията на движенията, дисметрия, скандирана реч и мозъчна асинергия.
Нарушенията на малкия мозък са основни и в посттравматичен синдром на Ман,което се характеризира с атаксия, дискоординация, асинергия, нистагъм. Травма или инфекция могат да причинят малкия мозък Синдром на Голдщайн-Райхман:нарушения на статиката и координацията на движенията, асинергия, преднамерен тремор, намален мускулен тонус, хиперметрия, мегалография, нарушено възприемане на масата (теглото) на обект в ръцете.
Нарушенията на мозъчната функция също могат да бъдат вродени по природа, като се проявяват по-специално, Синдром на Зееман:атаксия, забавено развитие на речта и впоследствие церебеларна дизартрия.
Вродена церебеларна атаксия Проявява се със забавяне на развитието на двигателните функции на детето (на 6-месечна възраст не може да седи, започва да ходи късно, докато походката е атактична), както и забавяне на говора, продължително запазване на дизартрия, понякога изостават в умственото развитие, а микрокраниалните прояви не са рядкост. При КТ мозъчните полукълба са намалени. До около 10-годишна възраст обикновено настъпва компенсация на мозъчните функции, които обаче могат да бъдат нарушени под въздействието на вредни екзогенни влияния. Възможни са и прогресиращи форми на заболяването.
Проява на вродена хипоплазия на малкия мозък е и Синдром на Фанкони-Търнър.Характеризира се с нарушена статика и координация на движенията, нистагъм, които обикновено са придружени от умствена изостаналост.
Вроденият включва и автозомно-рецесивен унаследен тип, който се среща рядко болест на Бетен:Характеризира се с вродена церебеларна атаксия, проявяваща се през първата година от живота с нарушена статика и координация на движенията, нистагъм, нарушение на координацията на погледа и умерена мускулна хипотония. Възможни са диспластични признаци. Детето закъснява, понякога само на 2-3 годишна възраст, започва да държи главата си, дори по-късно - да стои, да ходи, да говори. Речта му се променя според вида на церебеларната дизартрия. Възможни са вегетативно-висцерални нарушения, прояви на имуносупресия. След няколко години клиничната картина обикновено се стабилизира, пациентът до известна степен се адаптира към съществуващите дефекти.
Спастична атаксия по предложение на A. Bell и E. Carmichel (1939) е наречена церебеларна атаксия, наследена от автозомно доминантен тип, която се характеризира с началото на заболяването на 3-4-годишна възраст и се проявява с комбинация от мозъчен мозък. атаксия с дизартрия, хиперрефлексия на сухожилията и повишен мускулен тонус по спастичен тип, като е възможно (но не задължителни признаци на заболяването) атрофия на зрителните нерви, дегенерация на ретината, нистагъм, окуломоторни нарушения.
Автозомно доминантно се унаследява синдром на Фелдман(описано от немския лекар Х. Фелдман, роден през 1919 г.): церебеларна атаксия, преднамерен тремор и ранно побеляване на косата. Проявява се през второто десетилетие от живота и по-нататък бавно прогресира, което води до инвалидизация след 20-30 години.
Късна мозъчна атрофия или Синдром на Томописан през 1906 г. от френския невролог A. Thomas (1867-1963), обикновено се проявява при лица над 50-годишна възраст с прогресивна атрофия на кората на малкия мозък. Във фенотипа се появяват признаци на церебеларен синдром, предимно мозъчна статична и локомоторна атаксия, скандирана реч и промени в почерка. В далеч напреднал стадий са възможни прояви на пирамидална недостатъчност.
Комбинацията от мозъчни нарушения с миоклонус се характеризира с Хънт миоклонична церебеларна дисинергия,или миоклонус атаксия,с този симптомокомплекс в клиничната картина се проявяват умишлен тремор, миоклонус, възникващ в ръцете и впоследствие придобиващ генерализиран характер, атаксия и диссинергия, нистагъм, скандирана реч, понижен мускулен тонус. То е следствие от дегенерация на мозъчните ядра, червените ядра и техните връзки, както и кортикално-подкоровите структури.
В напреднал стадий на заболяването са възможни епилептични припадъци и деменция. Прогнозата е лоша. Отнася се за рядка форма на прогресираща наследствена атаксия. Наследява се по автозомно рецесивен начин. Обикновено се появява в млада възраст. Оспорва се нозологичната независимост на симптомокомплекса. Американският невролог Р. Хънт (1872-1937) описва заболяването през 1921г.
Сред дегенеративните процеси определено място заема дегенерация на малкия мозък на Холмс,или фамилна мозъчна атрофия,или прогресивна атрофия на церебеларната система, главно на зъбчатите ядра, както и червените ядра, докато проявите на демиелинизация се изразяват в горната мозъчна педикула. Характеризира се със статична и динамична атаксия, асинергия, нистагъм, дизартрия, намален мускулен тонус, мускулна дистония, тремор на главата, миоклонус. Епилептичните припадъци се появяват почти едновременно. Интелигентността обикновено се запазва. ЕЕГ показва пароксизмална аритмия. Заболяването се признава за наследствено, но видът на унаследяване не е посочен. Описано заболяването през 1907 г. от английския невропатолог Г. Холмс
(1876-1965).
Алкохолна дегенерация на малкия мозък - следствие от хронична алкохолна интоксикация. Засяга се главно мозъчният червей, като основно се проявява мозъчна атаксия и нарушена координация на движенията на краката, докато движенията на ръцете, окуломоторните и говорните функции са нарушени в много по-малка степен. Обикновено това заболяване е придружено от изразена загуба на паметта в комбинация с полиневропатия.
се проявява като церебеларна атаксия, която понякога може да бъде единственият клиничен симптом, свързан със злокачествен тумор, без локални признаци, показващи мястото на неговото възникване. Паранеопластична мозъчна дегенерацияможе да бъде по-специално вторична проява на рак на гърдата или на яйчниците.
Синдром на Баракер-Бордас-Руис-Лара се проявява като мозъчни нарушения, възникващи във връзка с бързо прогресираща атрофия на малкия мозък. Синдром при пациенти с рак на бронхите, придружен от обща интоксикация, е описан от съвременния испански лекар L. Barraquer-Bordas (роден през 1923 г.).
Рядко се среща рецесивна Х хромозомна атаксия- наследствено заболяване, което се проявява почти само при мъже с бавно прогресираща мозъчна недостатъчност. Предава се по рецесивен, свързан с пола тип.
Заслужава внимание и фамилна пароксизмална атаксия,или периодична атаксия.Дебютира по-често в детството, но може да се появи и по-късно – до 60 години. Клиничната картина се свежда до пароксизмални прояви на нистагъм, дизартрия и атаксия, понижен мускулен тонус, виене на свят, гадене, повръщане, главоболие, с продължителност от няколко минути до 4 седмици.
Пристъпите на фамилна пароксизмална атаксия могат да бъдат предизвикани от емоционален стрес, физическа умора, треска, прием на алкохол, докато между пристъпите фокални неврологични симптоми в повечето случаи не се откриват, но понякога са възможни нистагъм и леки мозъчни симптоми.
Морфологичният субстрат на заболяването се разпознава като атрофичен процес главно в предната част на мозъчния червей. За първи път заболяването е описано през 1946 г. от М. Паркър. Наследява се по автозомно доминантен начин. През 1987 г. при фамилна пароксизмална атаксия е установено намаляване на активността на пируват дехидрогеназата на кръвните левкоцити до 50-60% от нормалното ниво. През 1977 г. R. Lafrance et al. обърна внимание на високия профилактичен ефект на диакарб, по-късно флунаризин беше предложен за лечение на фамилна пароксизмална атаксия.
Остра церебеларна атаксия или синдром на Лайден-Вестфал,е добре дефиниран симптомокомплекс, който е параинфекциозно усложнение. По-често се среща при деца 1-2 седмици след обща инфекция (грип, тиф, салмонелоза и др.). Характеризира се с груба статична и динамична атаксия, преднамерен тремор, хиперметрия, асинергия, нистагъм, скандирана реч, понижен мускулен тонус. В цереброспиналната течност се открива лимфоцитна плеоцитоза, умерено повишаване на протеина. В началото на заболяването са възможни замайване, нарушения на съзнанието, конвулсии. При CT и MRI патология не се открива. Курсът е доброкачествен. В повечето случаи след няколко седмици или месеци - пълно възстановяване, понякога - остатъчни нарушения под формата на лека мозъчна недостатъчност.
Болестта на Мари-Фоа-Алахуанин - късна симетрична кортикална атрофия на малкия мозък с преобладаваща лезия на пириформни неврони (клетки на Пуркине) и зърнестия слой на кората, както и устната част на мозъчния вермис и маслинова дегенерация. Проявява се при лица на възраст 40-75 години с нарушение на равновесието, атаксия, нарушение на походката, нарушения на координацията и намален мускулен тонус, предимно в краката; умишленият тремор в ръцете не е много изразен. Възможни са говорни нарушения, но не принадлежат към задължителните признаци на заболяването. Заболяването е описано през 1922 г. от френските невропатолози P. Marie, Ch. Foix и Th. алажуанин. Заболяването е спорадично. Етиологията на заболяването не е изяснена. Има мнения за провокиращата роля на интоксикацията, преди всичко злоупотребата с алкохол, както и хипоксия, наследствена обремененост. Клиничната картина се потвърждава от КТ данни на главата, което разкрива изразено намаляване на обема на малкия мозък на фона на дифузни атрофични процеси в мозъка. В допълнение, високото ниво на аминотрансферазите в кръвната плазма се признава за характерно (Ponomareva E.N. et al., 1997).
