7.1. STRUCTURE, CONNEXIONS ET FONCTIONS DU CEREBELLE
Le cervelet (cervelet) est situé sous une dure-mère en double appelée contour du cervelet(tentorium cerebelli), qui divise la cavité crânienne en deux espaces inégaux - supratentorielle et subtentorielle. V espace sous-tentorielle, dont le fond est la fosse crânienne postérieure, en plus du cervelet, est le tronc cérébral. Le volume du cervelet est en moyenne de 162 cm3. Son poids varie entre 136 et 169 g.
Le cervelet est situé au-dessus du pont et de la moelle allongée. Avec les voiles cérébrales supérieure et inférieure, il constitue le toit du quatrième ventricule cérébral, dont le fond est la fosse dite rhomboïde (voir chapitre 9). Au-dessus du cervelet se trouvent les lobes occipitaux du gros cerveau, séparés de lui par la tente du cervelet.
Dans le cervelet, il y a deux hémisphères(hémispherum cervelli). Entre eux, dans le plan sagittal au-dessus du ventricule IV du cerveau, se trouve la partie phylogénétiquement la plus ancienne du cervelet - son Ver de terre(vermis du cervelet). Le vermis et les hémisphères cérébelleux sont fragmentés en lobules par de profonds sillons transversaux.
Le cervelet est composé de matière grise et blanche. La matière grise forme le cortex cérébelleux et les noyaux appariés noyaux cervelet situés dans sa profondeur (Fig. 7.1). Les plus grands d'entre eux sont grains dentelés(nucleus dentatus) - situé dans les hémisphères. Dans la partie centrale du ver, il y a noyaux de tente(noyaux
Riz. 7.1. Noyaux cérébelleux.
1 - noyau denté; 2 - noyau liégeux; 3 - le noyau de la tente; 4 - noyau sphérique.
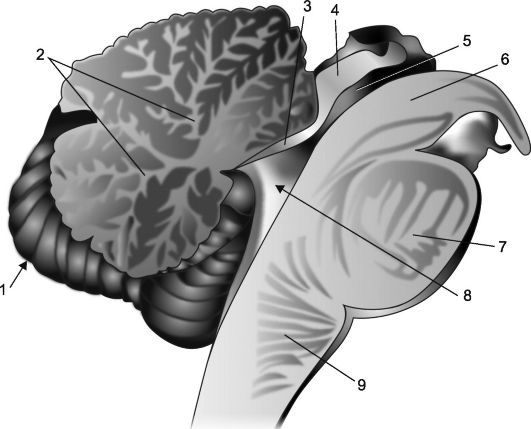
Riz. 7.2.Coupe sagittale du cervelet et du tronc cérébral.
1 - cervelet; 2 - "arbre de vie"; 3 - voile du cerveau antérieur; 4 - plaque du quadruple; 5 - aqueduc du cerveau; 6 - la jambe du cerveau; 7 - pont; 8 - Ventricule IV, son plexus choroïde et sa tente ; 9 - moelle allongée.
fastigii), entre eux et les noyaux dentés sont sphérique et noyaux liégeux(nuctei.globosus et emboliformis).
Du fait que le cortex recouvre toute la surface du cervelet et pénètre dans la profondeur de ses sillons, sur une coupe sagittale du cervelet, son tissu présente un motif foliaire dont les nervures sont formées par une substance blanche (Fig. . 7.2), qui constitue le soi-disant arbre de vie du cervelet (arbor vitae cervelli). À la base de l'arbre de vie, il y a une encoche en forme de coin, qui est la partie supérieure de la cavité du ventricule IV; les bords de cet évidement forment sa tente. Le toit de la tente est un ver cérébelleux et ses parois antérieure et postérieure sont de fines plaques cérébrales appelées antérieure et postérieure le cerveau navigue(vella médullaire antérieure et postérieure).
Quelques informations sur l'architectonique cérébelleuse, donnant des motifs pour juger de la fonction de ses composants. Ont cortex cérébelleux Il existe deux couches cellulaires : la couche interne est granulaire, constituée de petites cellules à grains, et la couche externe est moléculaire. Entre elles, il y a un certain nombre de grandes cellules en forme de poire portant le nom du scientifique tchèque I. Purkinje qui les a décrites (Purkinje I., 1787-1869).
Les impulsions pénètrent dans le cortex cérébelleux à travers des fibres moussues et rampantes qui y pénètrent à partir de la substance blanche, qui constituent les voies afférentes du cervelet. À travers les fibres moussues, les impulsions de la moelle épinière
les noyaux vestibulaires et les noyaux du pont sont transférés aux cellules de la couche granuleuse du cortex. Les axones de ces cellules, ainsi que les fibres rampantes traversant la couche granuleuse en transit et transportant des impulsions des olives inférieures au cervelet, atteignent la couche moléculaire superficielle du cervelet. Ici, les axones des cellules de la couche granuleuse et les fibres rampantes se divisent en forme de T et, dans la couche moléculaire, leurs branches prennent une direction longitudinale à la surface du cervelet. Les impulsions qui ont atteint la couche moléculaire du cortex, après avoir traversé les contacts synaptiques, tombent sur les dendrites ramifiées des cellules de Purkinje situées ici. Ensuite, ils suivent les dendrites des cellules de Purkinje jusqu'à leurs corps situés à la frontière des couches moléculaires et granulaires. Puis, le long des axones des mêmes cellules traversant la couche granuleuse, elles pénètrent dans les profondeurs de la substance blanche. Les axones des cellules de Purkinje se terminent dans les noyaux du cervelet. Principalement dans le noyau denté. Des influx efférents provenant du cervelet le long des axones des cellules qui composent son noyau et participant à la formation des pédoncules cérébelleux quittent le cervelet.
Le cervelet a trois paires de pattes : bas, milieu et haut. Le bas de la jambe le relie à la moelle allongée, le milieu - au pont, le haut - au mésencéphale. Les jambes du cerveau constituent les voies qui transportent les impulsions vers et depuis le cervelet.
Le ver cérébelleux assure la stabilisation du centre de gravité du corps, son équilibre, sa stabilité, la régulation du tonus des groupes musculaires réciproques, principalement le cou et le tronc, et l'émergence de synergies cérébelleuses physiologiques qui stabilisent l'équilibre du corps.
Pour maintenir avec succès l'équilibre du corps, le cervelet reçoit constamment des informations passant le long des voies spinocérébelleuses des propriocepteurs de diverses parties du corps, ainsi que des noyaux vestibulaires, des olives inférieures, de la formation réticulaire et d'autres formations impliquées dans le contrôle de la position du corps. pièces dans l'espace. La plupart des voies afférentes menant au cervelet passent par le pédicule cérébelleux inférieur, certaines d'entre elles sont situées dans le pédicule cérébelleux supérieur.
Impulsions de sensibilité proprioceptive, allant au cervelet, comme d'autres impulsions sensorielles, suivant les dendrites des premiers neurones sensoriels, atteignent leurs corps situés dans les ganglions rachidiens. Par la suite, les impulsions allant au cervelet le long des axones des mêmes neurones sont dirigées vers les corps des deuxièmes neurones, qui sont situés dans les parties internes de la base des cornes postérieures, formant ce que l'on appelle Les piliers de Clark. Leurs axones tombent dans les sections latérales des cordons latéraux de la moelle épinière, où ils forment voies spinocérébelleuses, dans ce cas, une partie des axones tombe dans la colonne latérale du même côté et s'y forme le tractus spinocérébelleux postérieur de Flexig (tractus spinocerebellaris postérieur). Une autre partie des axones des cellules des cornes postérieures passe de l'autre côté de la moelle épinière et pénètre dans la moelle latérale opposée, s'y formant voie spinocérébelleuse antérieure de Govers (tractus spinocerebellaris antérieur). Les faisceaux spinocérébelleux, de volume croissant au niveau de chaque segment rachidien, montent jusqu'au bulbe rachidien.
Dans la moelle allongée, la voie spinocérébelleuse postérieure dévie latéralement et, après avoir traversé le pédicule cérébelleux inférieur, pénètre dans le cervelet. La voie spinocérébelleuse antérieure passe en transit à travers la moelle allongée, le pont du cerveau et atteint le mésencéphale, au niveau duquel elle effectue sa deuxième traversée dans le velum cérébral antérieur et passe dans le cervelet par le pédoncule cérébelleux supérieur.
Ainsi, des deux faisceaux rachidiens, l'un n'est jamais traversé (chemin de Fleksig décroisé), et l'autre passe deux fois du côté opposé (traversé deux fois par le chemin de Govers). En conséquence, les deux conduisent des impulsions de chaque moitié du corps, principalement vers la moitié homolatérale du cervelet.
En plus des faisceaux spinocérébelleux de Fleksig, les impulsions vers le cervelet passent par le pédicule cérébelleux inférieur le long tractus vestibulo-cérébelleux (tractus vestibulocerebellaris), commençant principalement dans le noyau vestibulaire supérieur de la spondylarthrite ankylosante, et le long tractus olivomocérébelleux (tractus olivocerebellaris), provenant de l'olive inférieure. Une partie des axones des cellules des noyaux minces et en forme de coin, ne participant pas à la formation du tractus bulbo-thalamique, sous forme de fibres arquées externes (fiber arcuatae externae) pénètre également dans le cervelet par le pédoncule cérébelleux inférieur.
À travers ses pattes médianes, le cervelet reçoit des impulsions du cortex cérébral. Ces impulsions passent par voies cortico-cérébellopontines, constituées de deux neurones. Les corps des premiers neurones sont situés dans le cortex cérébral, principalement dans le cortex des parties postérieures des lobes frontaux. Leurs axones passent dans le cadre de la couronne radiante, la jambe antérieure de la capsule interne et se terminent dans les noyaux du pont. Axones des cellules des deuxièmes neurones, dont les corps sont situés dans leurs propres noyaux du pont, aller sur son côté opposé et maquiller, après l'intersection, le pédicule cérébelleux moyen,
se terminant dans l'hémisphère opposé du cervelet.
Certaines des impulsions apparues dans le cortex cérébral atteignent l'hémisphère opposé du cervelet, apportant des informations non pas sur le mouvement produit, mais uniquement sur le mouvement actif prévu. Ayant reçu de telles informations, le cervelet envoie instantanément des impulsions qui corrigent les mouvements volontaires, surtout, en éteignant l'inertie et le plus rationnel régulation du tonus musculaire réciproque - agonistes et antagonistes musculaires. En conséquence, une sorte de éimétrie, rendre les mouvements volontaires clairs, perfectionnés, dépourvus de composants inappropriés.
Les voies qui quittent le cervelet sont composées des axones des cellules, dont les corps forment ses noyaux. La plupart des voies efférentes, y compris les voies des noyaux dentés, quitter le cervelet par la partie supérieure de sa jambe. Au niveau des tubercules inférieurs du quadruple, le tractus cérébelleux efférent croise (intersection des pattes cérébelleuses supérieures de Werneking). Après avoir traversé chacun d'eux atteint les noyaux rouges du côté opposé du mésencéphale. Dans les noyaux rouges, les impulsions cérébelleuses passent au neurone suivant, puis se déplacent le long des axones des cellules dont les corps sont noyés dans les noyaux rouges. Ces axones sont formés dans voies rouges-nucléaires-rachidiennes (tracti rubro spinalis), les chemins de Monakov, qui peu après les sorties de grains rouges subissent une croix (croix de pneu ou croix de truite), après quoi ils descendent dans la moelle épinière. Dans la moelle épinière, les voies rachidiennes nucléaires rouges sont situées dans les moelles latérales; leurs fibres constitutives se terminent aux cellules des cornes antérieures de la moelle épinière.
L'ensemble de la voie efférente du cervelet aux cellules des cornes antérieures de la moelle épinière peut être appelé cérébelleux-rouge-nucléaire-spinal (tractus cerebello-rubrospinalis). Il traverse deux fois (intersection des pédoncules cérébelleux supérieurs et intersection de l'opercule) et relie finalement chaque hémisphère du cervelet aux motoneurones périphériques situés dans les cornes antérieures de la moitié homolatérale de la moelle épinière.
Depuis les noyaux du vermis cérébelleux, les voies efférentes passent principalement par le pédicule cérébelleux inférieur jusqu'à la formation réticulaire du tronc cérébral et des noyaux vestibulaires. De là, le long des voies réticulo-spinales et vestibulo-spinales passant le long des moelles antérieures de la moelle épinière, ils atteignent également les cellules des cornes antérieures. Une partie des impulsions provenant du cervelet, passant par les noyaux vestibulaires, pénètre dans le faisceau longitudinal médial, atteint les noyaux III, IV et VI des nerfs crâniens qui assurent le mouvement des globes oculaires et affecte leur fonction.
Pour résumer, il faut souligner les points suivants :
1. Chaque moitié du cervelet reçoit des impulsions principalement a) de la moitié homolatérale du corps, b) de l'hémisphère opposé du cerveau, qui a des connexions cortico-spinales avec la même moitié du corps.
(2) De chaque moitié du cervelet, des impulsions efférentes sont dirigées vers les cellules des cornes antérieures de la moitié homolatérale de la moelle épinière et vers les noyaux des nerfs crâniens qui assurent le mouvement des globes oculaires.
Cette nature des connexions cérébelleuses permet de comprendre pourquoi, lorsqu'une moitié du cervelet est touchée, les troubles cérébelleux surviennent principalement dans le même, c'est-à-dire. homolatéral, moitié du corps. Ceci est particulièrement prononcé lorsque les hémisphères cérébelleux sont touchés.
7.2. RECHERCHE DES FONCTIONS DU CEREBELLE
ET MANIFESTATIONS CLINIQUES DE SES DÉFAITES
Avec des dommages au cervelet, des troubles de la statique et de la coordination des mouvements, une hypotonie musculaire et un nystagmus sont caractéristiques.
Lésion cérébelleuse d'abord son ver, conduit à des violations de la statique - la capacité de maintenir une position stable du centre de gravité du corps humain, l'équilibre, la stabilité. Lorsque cette fonction est perturbée, ataxie statique (du grec ataxie - désordre, instabilité). L'instabilité du patient est notée. Par conséquent, en position debout, il écarte largement les jambes, équilibre avec ses mains. Une ataxie particulièrement clairement statique est détectée avec une diminution artificielle de la zone d'appui, en particulier dans la pose de Romberg. Le patient est invité à se lever, en bougeant fermement ses pieds et en relevant légèrement la tête. En présence de troubles cérébelleux, on note l'instabilité du patient dans cette position, son corps se balance, parfois il « tire » dans une certaine direction, et si le patient n'est pas soutenu, il peut tomber. En cas de lésion du ver cérébelleux, le patient se balance généralement d'un côté à l'autre et retombe souvent. Avec la pathologie de l'hémisphère cérébelleux, il y a une tendance à tomber principalement vers le foyer pathologique. Si le trouble statique est modérément exprimé, il est plus facile de l'identifier dans ce qu'on appelle compliqué ou pose Romberg sensibilisée. Le patient est invité à mettre ses pieds dans une ligne de sorte que l'orteil d'un pied repose sur le talon de l'autre. L'évaluation de la stabilité est la même que dans la position habituelle de Romberg.
Normalement, lorsqu'une personne est debout, les muscles de ses jambes sont tendus. (réaction de soutien), avec la menace de tomber sur le côté, sa jambe de ce côté se déplace dans la même direction et l'autre jambe se détache du sol (réaction de saut). Avec des dommages au cervelet (principalement le ver), les réactions du patient sont perturbées
soutenir et sauter. La violation de la réaction d'appui se manifeste par l'instabilité du patient en position debout, notamment en position de Romberg. La violation de la réaction de saut conduit au fait que si le médecin, se tenant derrière le patient et l'assurant, pousse le patient dans un sens ou dans l'autre, alors le patient tombe avec une légère poussée (symptôme de poussée).
En cas de lésion du cervelet, la démarche du patient est généralement modifiée en raison du développement ataxie statolokomotrice. Démarche cérébelleuse ressemble à bien des égards à la démarche d'une personne ivre, c'est pourquoi on l'appelle parfois la « démarche d'un ivrogne ». En raison de l'instabilité, le patient marche de manière incertaine, écartant largement les jambes, tandis qu'il est "jeté" d'un côté à l'autre. Et lorsque l'hémisphère cérébelleux est endommagé, il dévie en marchant d'une direction donnée vers le foyer pathologique. L'instabilité est particulièrement prononcée dans les virages. Si l'ataxie est prononcée, les patients perdent complètement la capacité de contrôler leur corps et peuvent non seulement se tenir debout et marcher, mais même s'asseoir.
La lésion prédominante des hémisphères cérébelleux conduit à un trouble de ses effets anti-inertiels, notamment à l'émergence ataxie cinétique. Elle se manifeste par la maladresse des mouvements et est particulièrement prononcée avec les mouvements qui demandent de la précision. Des tests de coordination des mouvements sont effectués pour détecter l'ataxie cinétique. Certains d'entre eux sont décrits ci-dessous.
Test de diadochokinèse (du grec diadochos - séquence). Le patient est invité à fermer les yeux, à tendre les bras en avant et rapidement, à supination rythmique et à pénétrer les mains. En cas de lésion de l'hémisphère cérébelleux, les mouvements de la main du côté du processus pathologique s'avèrent plus amples (conséquence de la dysmétrie, plus précisément de l'hypermétrie), en conséquence, la main commence à prendre du retard. Cela indique la présence d'adiadochokinèse.
Test au doigt. Un patient aux yeux fermés doit retirer sa main, puis, lentement, avec son index, toucher le bout du nez. En cas de pathologie cérébelleuse, la main du côté du foyer pathologique effectue un mouvement excessif de volume (hypermétrie),à la suite de quoi le patient manque. Le test au doigt révèle une caractéristique de la pathologie cérébelleuse tremblements cérébelleux (intentionnels), dont l'amplitude augmente à mesure que le doigt s'approche de la cible. Ce test vous permet d'identifier la soi-disant bradytélékinésie. (symptôme de bride): près de la cible, le mouvement du doigt ralentit, parfois même s'interrompt, puis reprend.
Test doigt-doigt. Un patient aux yeux fermés est invité à écarter largement les bras puis à rapprocher les index en essayant d'introduire le doigt dans le doigt, tandis que, comme pour le test du doigt, un tremblement intentionnel et un symptôme de bride sont révélés.
Test du genou calcanéen (fig. 7.3). Le patient, allongé sur le dos, les yeux fermés, est invité à lever une jambe en hauteur puis à frapper le genou de l'autre jambe avec son talon. Avec la pathologie cérébelleuse, le patient ne peut pas ou il lui est difficile d'introduire son talon dans le genou de l'autre jambe, en particulier lors d'un test avec la jambe homolatérale à l'hémisphère cérébelleux affecté. Si, néanmoins, le talon atteint le genou, il est alors proposé de le tenir en touchant légèrement la surface avant du bas de la jambe, jusqu'à l'articulation de la cheville, tandis qu'en cas de pathologie cérébelleuse, le talon glisse du bas de la jambe tout le temps dans un sens ou dans l'autre.

Riz. 7.3.Test du genou calcanéen.
Essai indicatif : Le patient est invité à frapper plusieurs fois la pointe en caoutchouc du marteau, qui est dans la main de l'examinateur, avec son index. En cas de pathologie cérébelleuse de la main du patient du côté de l'hémisphère cérébelleux affecté, il existe une chute mimique due à une dysmétrie.
Symptôme de Tom-Jumenti : Si le patient ramasse un objet, comme un verre, il écarte excessivement les doigts.
Nystagmus cérébelleux. La contraction des globes oculaires en regardant sur les côtés (nystagmus horizontal) est considérée comme une conséquence du tremblement intentionnel des globes oculaires (voir chapitre 30).
Trouble de la parole : La parole perd de sa fluidité, devient explosive, fragmentée, chantée comme une dysarthrie cérébelleuse (voir chapitre 25).
Modification de l'écriture manuscrite : En raison du trouble de la coordination des mouvements de la main, l'écriture devient inégale, les lettres sont déformées, trop grandes (mégalographie).
Phénomène pronatoire : Le patient est invité à garder les bras étendus vers l'avant en position de supination, tandis que la pronation spontanée se produit bientôt du côté de l'hémisphère cérébelleux affecté.
Symptôme de Hoff-Schilder : Si le patient tient ses bras tendus vers l'avant, alors du côté de l'hémisphère affecté, le bras est bientôt rétracté vers l'extérieur.
Un phénomène d'imitation. Un patient aux yeux fermés doit rapidement donner à la main une position similaire à celle que l'examinateur avait précédemment donnée à son autre main. Lorsque l'hémisphère cérébelleux est endommagé, la main homolatérale effectue un mouvement d'amplitude excessive.
Le phénomène de Doinikov. Phénomène de doigt. Le patient assis est invité à mettre les mains en supination avec les doigts écartés sur ses cuisses et à fermer les yeux. Dans le cas d'une lésion du cervelet du côté du foyer pathologique, une flexion spontanée des doigts et une pronation de la main et de l'avant-bras surviennent rapidement.
Symptôme de Stuart Holmes. L'examinateur demande au patient assis sur la chaise de plier les avant-bras en supination et en même temps, lui prenant les mains par les poignets, lui résiste. Si, en même temps, les mains du patient sont soudainement relâchées, la main du côté affecté, pliée par inertie, le frappera avec force dans la poitrine.
Hypotension musculaire. La défaite du vermis cérébelleux conduit généralement à une hypotension musculaire diffuse. Avec la défaite de l'hémisphère cérébelleux, les mouvements passifs révèlent une diminution du tonus musculaire du côté du processus pathologique. L'hypotonie musculaire entraîne la possibilité d'une hyperextension de l'avant-bras et du bas de la jambe (symptôme Olshansky) avec des mouvements passifs, à l'apparence symptômes d'une main ou d'un pied qui pendent quand ils sont secoués passivement.
Asynergies cérébelleuses pathologiques. Des violations des synergies physiologiques lors d'actes moteurs complexes sont révélées notamment lors des tests suivants (Fig. 7.4).
1. Asynergie selon Babinsky en position debout. Si un patient debout avec les jambes déplacées essaie de se pencher en arrière, en jetant la tête en arrière, alors normalement dans ce cas, une flexion des articulations du genou se produit. Dans la pathologie cérébelleuse due à l'asynergie, ce mouvement amical est absent, et le patient, en perte d'équilibre, retombe.
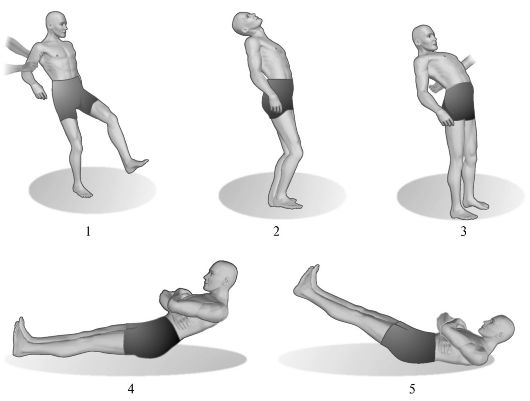
Riz. 7.4.Asynergie cérébelleuse.
1 - démarche d'un patient atteint d'ataxie cérébelleuse sévère; 2 - l'inclinaison arrière du corps est normale; 3 - avec des dommages au cervelet, le patient, se penchant en arrière, ne peut pas maintenir l'équilibre; 4 - effectuer un test d'asynergie cérébelleuse selon Babinsky par une personne en bonne santé; 5 - effectuer le même test chez les patients présentant des lésions cérébelleuses.
2. Asynergie selon Babinsky en décubitus dorsal. Le patient, allongé sur un plan ferme, les jambes tendues, écartées à la largeur de la ceinture scapulaire, est invité à croiser les bras sur sa poitrine puis à s'asseoir. En présence de pathologie cérébelleuse due à l'absence de contraction amicale des muscles fessiers (manifestation d'asynergie), le patient ne peut pas fixer les jambes et le bassin sur la zone d'appui, de ce fait, les jambes se soulèvent et il ne peut pas s'asseoir. L'importance de ce symptôme ne doit pas être surestimée chez les patients âgés, chez les personnes ayant une paroi abdominale flasque ou obèse.
En résumant ce qui précède, il convient de souligner la diversité et l'importance des fonctions exercées par le cervelet. Dans le cadre d'un mécanisme complexe de régulation de la rétroaction, le cervelet agit comme un point focal pour équilibrer le corps et maintenir le tonus musculaire. Comme le note P. Duus (1995), le cervelet permet d'effectuer des mouvements discrets et précis, De plus, l'auteur pense raisonnablement que le cervelet fonctionne comme un ordinateur, suivant et coordonnant les informations sensorielles à l'entrée et simulant les signaux moteurs à la sortie.
7.3. DÉGÉNÉRATIONS MULTISYSTÉMIQUES
Avec des signes de pathologie cérébelleuse
Les dégénérescences multisystémiques sont un groupe de maladies neurodégénératives dont la caractéristique commune est la nature multifocale de la lésion avec l'implication de divers systèmes fonctionnels et neurotransmetteurs du cerveau dans le processus pathologique et, par conséquent, la nature polysystémique des manifestations cliniques.
7.3.1. Ataxie cérébelleuse
Les ataxies spinocérébelleuses comprennent les maladies dégénératives héréditaires progressives, dans lesquelles les structures du cervelet, le tronc cérébral et les voies de la moelle épinière, qui sont principalement liées au système extrapyramidal, sont principalement affectées.
7.3.1.1. Ataxie héréditaire de Friedreich
Maladie héréditaire décrite en 1861 par le neuropathologiste allemand N. Friedreich (Friedreich N., 1825-1882). Elle se transmet sur le mode autosomique récessif ou (moins souvent) sur le mode autosomique dominant avec une pénétrance incomplète et une expression génique variable. Des cas sporadiques de la maladie sont également possibles.
Pathogénèsemaladie non précisée. En particulier, il n'y a aucune idée du défaut biochimique primaire qui en constitue la base.
Pathomorphologie.Les études pathologiques révèlent un amincissement prononcé de la moelle épinière en raison de processus atrophiques dans ses cordons postérieur et latéral. En règle générale, les voies cunéiformes (Burdach) et douces (Gaulle) et les voies spino-cérébelleuses de Govers et Fleksig souffrent, ainsi que la voie pyramidale croisée contenant
de nombreuses fibres liées au système extrapyramidal. Les processus dégénératifs sont également exprimés dans le cervelet, dans sa substance blanche et son appareil nucléaire.
Manifestations cliniques. La maladie se manifeste chez les enfants ou les jeunes de moins de 25 ans. S.N. Davidenkov (1880-1961) a noté que les signes cliniques de la maladie surviennent plus souvent chez les enfants de 6 à 10 ans. Le premier signe de maladie est généralement l'ataxie. Les patients éprouvent de l'incertitude, des titubations lors de la marche, des changements de démarche (lors de la marche, les jambes sont bien écartées). La démarche dans la maladie de Friedreich peut être appelée tabétique-cérébelleuse, car ses modifications sont causées par une combinaison d'ataxie sensible et cérébelleuse, ainsi que par une diminution généralement prononcée du tonus musculaire. Des troubles de la statique, une discoordination dans les mains, des tremblements intentionnels, une dysarthrie sont également caractéristiques. Nystagmus possible, surdité, éléments de la psalmodie, signes d'insuffisance pyramidale (hyperréflexie tendineuse, réflexes pathologiques du pied, parfois légère augmentation du tonus musculaire), besoin impérieux d'uriner, diminution de la puissance sexuelle. Parfois, une hyperkinésie athétoïde apparaît.
Un trouble précoce de la sensibilité profonde entraîne une diminution progressive des réflexes tendineux : d'abord sur les jambes, puis sur les bras. Au fil du temps, une hypotrophie musculaire des jambes distales se développe. La présence d'anomalies dans le développement du squelette est caractéristique. Tout d'abord, cela se manifeste par la présence Les pieds de Friedreich : le pied est raccourci, "creux", avec une cambrure très haute. Les phalanges principales de ses doigts ne sont pas pliées, les autres sont pliées (Fig. 7.5). Déformation possible de la colonne vertébrale, de la poitrine. Il existe souvent des manifestations de cardiopathie. La maladie progresse lentement, mais conduit régulièrement à l'invalidité des patients qui finissent par devenir alités.
Traitement. Le traitement pathogénétique n'a pas été développé. Prescrire des médicaments qui améliorent le métabolisme dans les structures du système nerveux, des agents fortifiants. En cas de déformation sévère des pieds, des chaussures orthopédiques sont indiquées.
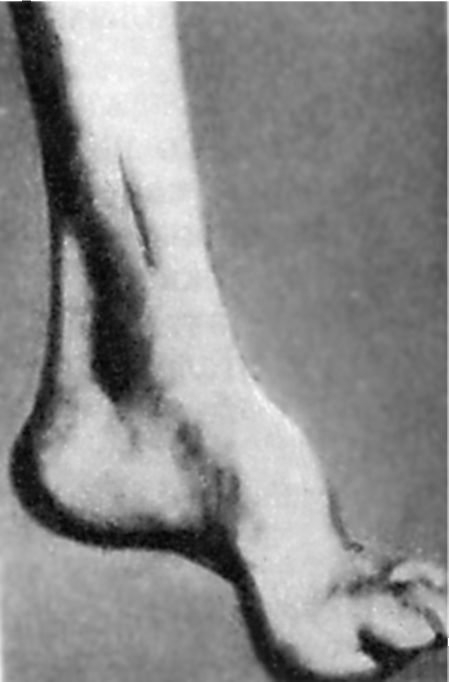
Riz. 7.5.Le pied de Friedreich.
7.3.1.2. Ataxie cérébelleuse héréditaire (maladie de Pierre Marie)
Il s'agit d'une maladie héréditaire chronique évolutive, qui se manifeste à l'âge de 30-45 ans, avec des troubles cérébelleux à croissance lente associés à des signes d'insuffisance pyramidale, tandis qu'une ataxie cérébelleuse statique et dynamique, des tremblements intentionnels, un discours chanté, une hyperréflexie tendineuse sont caractéristiques. Clonus possibles, réflexes pyramidaux pathologiques, strabisme, diminution de la vision, rétrécissement des champs visuels par atrophie primaire des nerfs optiques et dégénérescence pigmentaire de la rétine. L'évolution de la maladie progresse lentement. Il y a une diminution de la taille du cervelet, une dégénérescence cellulaire
Purkinje, olives inférieures, voies spinocérébelleuses. Il se transmet sur le mode autosomique dominant. La maladie a été décrite en 1893 par le neuropathologiste français R. Marie (1853-1940).
Actuellement, il n'y a pas unanimité dans la compréhension du terme « maladie de Pierre Marie », et la question de la possibilité de la séparer en une forme nosologique indépendante est discutable.
Aucun traitement n'a été développé. Un tonique général et métaboliquement actif, ainsi que des agents symptomatiques sont généralement utilisés.
7.3.2. Dystrophie olivopontocérébelleuse (maladie de Dejerine-Thom)
Il s'agit d'un groupe de maladies héréditaires chroniques évolutives dans lesquelles des modifications dystrophiques se développent principalement dans le cervelet, les olives inférieures, les noyaux du pont et les structures cérébrales associées.
Avec le développement de la maladie à un jeune âge, environ la moitié des cas sont hérités de manière dominante ou récessive, le reste est sporadique. Dans les cas sporadiques de la maladie, les manifestations du syndrome akinétique-rigide et de l'insuffisance autonome progressive sont plus fréquentes. L'âge moyen du patient présentant la manifestation de la forme héréditaire de la maladie dans le phénotype est de 28 ans, avec le sporadique - 49 ans, l'espérance de vie moyenne est de 14,9 et 6,3 ans, respectivement. Sous forme sporadique, en plus de l'atrophie des olives, du pont et du cervelet, on trouve plus souvent des lésions des cordons latéraux de la moelle épinière, de la substance noire et du striatum, une tache bleuâtre dans la fosse rhomboïde du quatrième ventricule cérébral.
Les symptômes du syndrome cérébelleux croissant sont caractéristiques. Troubles possibles de la sensibilité, éléments de syndromes bulbaires et akinétiques-rigides, hyperkinésie, en particulier myorrythmies de la langue et du voile du palais, ophtalmoparésie, diminution de l'acuité visuelle, troubles intellectuels. La maladie a été décrite en 1900 par les neuropathologistes français J. Dejerine et A. Thomas.
La maladie débute souvent par des troubles de la marche - instabilité, discoordination, chutes inattendues sont possibles. Ces troubles peuvent être la seule manifestation de la maladie dans un délai de 1 à 2 ans. À l'avenir, des troubles de la coordination apparaissent et se développent dans les mains: les manipulations avec de petits objets sont difficiles, l'écriture manuscrite est perturbée, un tremblement intentionnel se produit. La parole devient intermittente, floue, avec une teinte nasale et un rythme respiratoire qui ne correspond pas à la structure de la parole (le patient parle comme s'il était étranglé). A ce stade de la maladie, des manifestations d'insuffisance autonome progressive se rejoignent, des signes de syndrome akinétique-rigide apparaissent. Parfois, les symptômes dominants pour le patient sont la dysphagie, des crises d'étouffement nocturne. Ils se développent en relation avec une parésie mixte des muscles bulbaires et peuvent mettre la vie en danger.
En 1970, les neuropathologistes allemands B.W. Königsmark et L.P. Weiner distingué 5 types principaux dystrophie olivopontocérébelleuse, différant soit par les manifestations cliniques et morphologiques, soit par le type de transmission.
je type (type Menzel). À l'âge de 14-70 ans (plus souvent de 30 à 40 ans), il se manifeste par une ataxie, une dysarthrie, une dysphonie, une hypotonie musculaire, à un stade avancé - un tremblement brutal de la tête, du tronc, des bras, des muscles, des signes d'akinétique- syndrome rigide. Signes pyramidaux pathologiques possibles, parésie du regard, ophtalmoplégie externe et interne, troubles de la sensibilité, démence. Il se transmet sur le mode autosomique dominant. Elle a été distinguée comme forme indépendante en 1891 par P. Menzel.
II type (type Fickler-Winkler) ... A l'âge de 20-80 ans, il se manifeste par une ataxie, une diminution du tonus musculaire et des réflexes tendineux. Elle se transmet sur le mode autosomique récessif. Des cas sporadiques sont possibles.
III type avec dégénérescence rétinienne. Elle se manifeste dans l'enfance ou le jeune âge (jusqu'à 35 ans) : ataxie, tremblements de la tête et des extrémités, dysarthrie, signes d'insuffisance pyramidale, diminution progressive de la vision avec aboutissement à la cécité ; nystagmus possible, ophtalmoplégie, troubles sensitifs parfois dissociés. Il se transmet sur le mode autosomique dominant.
IV type (type Bouffon-Highmaker). À l'âge de 17-30 ans, il débute avec une ataxie cérébelleuse ou des signes de paraparésie spastique inférieure, dans les deux cas, déjà au stade précoce de la maladie, une combinaison de ces manifestations se forme, à laquelle des éléments de syndrome bulbaire, de parésie de les muscles faciaux, et des troubles de sensibilité profonde sont ajoutés par la suite. Dominant hérité.
V Type de. Elle se manifeste à l'âge de 7-45 ans : ataxie, dysarthrie, signes de syndrome akinétique-rigide et autres troubles extrapyramidaux, ophtalmoplégie progressive et démence sont possibles. Dominant hérité.
7.3.3. Dégénérescence olivorubrocérébelleuse (syndrome de Lejeune-Lermitte, maladie de Lermitte)
La maladie est caractérisée par une atrophie progressive du cervelet, principalement de son cortex, des noyaux dentés et des pédoncules cérébelleux supérieurs, des olives inférieures et des noyaux rouges. Il se manifeste principalement par une ataxie statique et dynamique. À l'avenir, d'autres signes de syndrome cérébelleux et de lésions du tronc cérébral sont possibles. La maladie a été décrite par les neuropathologistes français J. Lhermitte (Lhermitte J.J., 1877-1959) et J. Lezhon (Lejonne J., né en 1894).
7.3.4. Atrophie multisystémique
Au cours des dernières décennies, une maladie neurodégénérative sporadique et progressive appelée atrophie multisystémique a été identifiée comme une forme indépendante. Elle se caractérise par une lésion combinée des noyaux gris centraux, du cervelet, du tronc cérébral et de la moelle épinière. Les principales manifestations cliniques : parkinsonisme, ataxie cérébelleuse, signes d'insuffisance pyramidale et autonome (Levin O.S., 2002). Selon la prédominance de certaines caractéristiques du tableau clinique, on distingue trois types d'atrophie multisystémique.
1) de type olivopontocérébelleux, caractérisé par la prédominance des signes d'atteinte cérébelleuse ;
2) type strionigral, dans lequel les signes de parkinsonisme dominent;
3) Le syndrome de Shay-Drager, caractérisé par la prédominance des signes d'insuffisance autonome progressive avec des symptômes d'hypotension artérielle orthostatique dans le tableau clinique.
La base de l'atrophie multisystémique est la dégénérescence sélective de certaines zones de la matière principalement grise du cerveau avec des dommages aux neurones et aux éléments gliaux. Les causes des manifestations dégénératives dans le tissu cérébral restent inconnues aujourd'hui. Les manifestations d'atrophie multisystémique de type olivopontocérébelleux sont associées à des dommages aux cellules de Purkinje dans le cortex cérébelleux, ainsi qu'aux neurones des olives inférieures, aux noyaux pontocérébelleux, à la démyélinisation et à la dégénérescence, principalement du tractus de conduction pontocérébelleux.
Les troubles cérébelleux sont généralement une ataxie statique et dynamique avec altération des mouvements locomoteurs. Caractérisé par une instabilité en position de Romberg, une ataxie lors de la marche, une dysmétrie, une adiadocokinésie, un tremblement intentionnel, il peut y avoir un nystagmus (horizontal vertical, coups vers le bas), des mouvements du regard intermittents et lents, une altération de la convergence des yeux, un discours chanté.
L'atrophie multisystémique survient généralement à l'âge adulte et progresse rapidement. Le diagnostic repose sur des données cliniques et se caractérise par une combinaison de signes de parkinsonisme, d'insuffisance cérébelleuse et de troubles végétatifs. Le traitement de la maladie n'a pas été développé. La durée de la maladie - dans les 10 ans, se termine par la mort.
7.4. AUTRES MALADIES ASSOCIÉES AUX SIGNES DE LÉSIONS CÉRÉBRALES
Si le patient présente des signes de lésion cérébelleuse, alors dans la plupart des cas, tout d'abord vous devez penser à la possibilité tumeurs cérébelleuses(astrocytome, angioblastome, médulloblastome, tumeurs métastatiques) ou la sclérose en plaques. À tumeurs cérébelleuses les premiers signes d'hypertension intracrânienne apparaissent. Dans la sclérose en plaques, il est généralement possible d'identifier, en plus de la pathologie cérébelleuse, des manifestations cliniques de lésions d'autres structures du système nerveux central, principalement les systèmes visuel et pyramidal. En neurologie classique, la caractéristique de sclérose en plaque Triade de Charcot : nystagmus, tremblement intentionnel et parole chantée, et Syndrome de Nonne : trouble de la coordination des mouvements, dysmétrie, discours chanté et asynergies cérébelleuses.
Les troubles cérébelleux sont majeurs et en syndrome de Mann post-traumatique, qui se caractérise par une ataxie, une discoordination, une asynergie, un nystagmus. Un traumatisme ou une infection peut provoquer des lésions cérébelleuses Syndrome de Goldstein-Reichmann : troubles de la statique et de la coordination des mouvements, asynergie, tremblements intentionnels, diminution du tonus musculaire, hypermétrie, mégalographie, altération de la perception de la masse (poids) d'un objet dans les mains.
Les troubles de la fonction cérébelleuse peuvent également être de nature congénitale, se manifestant notamment par Syndrome de Zeeman : ataxie, retard du développement de la parole et par la suite dysarthrie cérébelleuse.
Ataxie cérébelleuse congénitale Il se manifeste par un retard dans le développement des fonctions motrices de l'enfant (à l'âge de 6 mois, il ne peut pas s'asseoir, il commence à marcher tard, alors que la démarche est atactique), ainsi qu'un retard de la parole, une préservation prolongée de la dysarthrie, parfois le retard mental et les manifestations microcrâniennes ne sont pas rares. Au scanner, les hémisphères cérébelleux sont réduits. Vers l'âge de 10 ans environ, il se produit généralement une compensation des fonctions cérébrales, qui peut cependant être perturbée sous l'influence d'influences exogènes néfastes. Des formes évolutives de la maladie sont également possibles.
Une manifestation d'hypoplasie congénitale du cervelet est et Syndrome de Fancony-Turner. Elle se caractérise par une altération de la statique et de la coordination des mouvements, le nystagmus, qui s'accompagne généralement d'un retard mental.
Congénital comprend également un type héréditaire autosomique récessif, qui est rarement trouvé Maladie de Betten : Elle se caractérise par une ataxie cérébelleuse congénitale, qui se manifeste au cours de la première année de vie par une altération de la statique et de la coordination des mouvements, un nystagmus, un trouble de la coordination du regard et une hypotonie musculaire modérée. Des signes dysplasiques sont possibles. L'enfant est en retard, parfois seulement à 2-3 ans, commence à se tenir la tête, même plus tard - se tenir debout, marcher, parler. Son discours est modifié selon le type de dysarthrie cérébelleuse. Troubles végétatifs-viscéraux possibles, manifestations d'immunosuppression. Après quelques années, le tableau clinique se stabilise généralement, le patient s'adapte dans une certaine mesure aux défauts existants.
Ataxie spastique à la suggestion de A. Bell et E. Carmichel (1939), l'ataxie cérébelleuse héritée d'un type autosomique dominant a été nommée, qui se caractérise par l'apparition de la maladie à 3-4 ans et se manifeste par une combinaison de ataxie avec dysarthrie, hyperréflexie tendineuse et augmentation du tonus musculaire par type spastique, alors que possible (mais non signe obligatoire de la maladie) atrophie des nerfs optiques, dégénérescence rétinienne, nystagmus, troubles oculomoteurs.
L'autosomique dominante est héréditaire syndrome de Feldman(décrit par le médecin allemand H. Feldmann, né en 1919) : ataxie cérébelleuse, tremblements intentionnels et grisonnement précoce des cheveux. Elle se manifeste dans la deuxième décennie de la vie et progresse lentement, conduisant à un handicap dans 20-30 ans.
Atrophie cérébelleuse tardive ou Le syndrome de Tom décrit en 1906 par le neurologue français A. Thomas (1867-1963), se manifeste généralement chez les personnes de plus de 50 ans avec une atrophie progressive du cortex cérébelleux. Dans le phénotype, des signes de syndrome cérébelleux apparaissent, principalement une ataxie statique et locomotrice cérébelleuse, un discours chanté et des modifications de l'écriture. A un stade avancé, des manifestations d'insuffisance pyramidale sont possibles.
L'association de troubles cérébelleux et de myoclonies se caractérise par Chasse à la dyssynergie myoclonique cérébelleuse, ou myoclonie-ataxie, avec ce complexe de symptômes dans le tableau clinique, se manifestent des tremblements intentionnels, des myoclonies apparaissant dans les mains et acquérant plus tard un caractère généralisé, une ataxie et une dyssynergie, un nystagmus, un discours chanté, une diminution du tonus musculaire. C'est une conséquence de la dégénérescence des noyaux cérébelleux, des noyaux rouges et de leurs connexions, ainsi que des structures cortico-sous-corticales.
À un stade avancé de la maladie, des crises d'épilepsie et une démence sont possibles. Le pronostic est mauvais. Désigne une forme rare d'ataxie héréditaire progressive. Elle se transmet sur le mode autosomique récessif. Il apparaît généralement à un jeune âge. L'indépendance nosologique du complexe symptomatique est contestée. Le neurologue américain R. Hunt (1872-1937) a décrit la maladie en 1921.
Parmi les processus dégénératifs, une certaine place est occupée par dégénérescence cérébelleuse de Holmes, ou atrophie cérébelleuse familiale, ou atrophie progressive du système cérébelleux, principalement des noyaux dentés, ainsi que des noyaux rouges, tandis que des manifestations de démyélinisation s'expriment dans le pédicule cérébelleux supérieur. Caractérisé par une ataxie statique et dynamique, une asynergie, un nystagmus, une dysarthrie, une diminution du tonus musculaire, une dystonie musculaire, des tremblements de la tête, des myoclonies. Les crises d'épilepsie apparaissent presque simultanément. L'intelligence est généralement préservée. L'EEG montre une dysrythmie paroxystique. La maladie est reconnue comme héréditaire, mais le type de transmission n'est pas précisé. Décrit la maladie en 1907 par le neuropathologiste anglais G. Holmes
(1876-1965).
Dégénérescence cérébelleuse alcoolique - une conséquence d'une intoxication chronique à l'alcool. Le ver cérébelleux est principalement affecté, avec une ataxie cérébelleuse et une altération de la coordination des mouvements des jambes se manifestant principalement, tandis que les mouvements de la main, les fonctions oculomotrices et la parole sont altérés dans une bien moindre mesure. Habituellement, cette maladie s'accompagne d'une perte de mémoire prononcée associée à une polyneuropathie.
se manifeste par une ataxie cérébelleuse, qui peut parfois être le seul symptôme clinique associé à une tumeur maligne, sans signes locaux indiquant le lieu de sa survenue. Dégénérescence cérébelleuse paranéoplasique peut être notamment une manifestation secondaire d'un cancer du sein ou de l'ovaire.
Syndrome de Barraquer-Bordas-Ruiz-Lara se manifeste par des troubles cérébelleux résultant d'une atrophie cérébelleuse rapidement progressive. Un syndrome chez les patients atteints de cancer bronchique accompagné d'une intoxication générale est décrit par le médecin espagnol moderne L. Barraquer-Bordas (né en 1923).
Rarement trouvé ataxie chromosomique X récessive- une maladie héréditaire qui se manifeste presque exclusivement chez l'homme présentant une insuffisance cérébelleuse lentement évolutive. Elle est transmise dans un type récessif, lié au sexe.
Remarquable et ataxie paroxystique familiale, ou ataxie périodique. Il fait ses débuts plus souvent dans l'enfance, mais il peut également apparaître plus tard - jusqu'à 60 ans. Le tableau clinique est réduit à des manifestations paroxystiques de nystagmus, dysarthrie et ataxie, diminution du tonus musculaire, vertiges, nausées, vomissements, maux de tête, durant de quelques minutes à 4 semaines.
Les crises d'ataxie paroxystique familiale peuvent être déclenchées par un stress émotionnel, une fatigue physique, de la fièvre, une consommation d'alcool, tandis qu'entre les crises, les symptômes neurologiques focaux dans la plupart des cas ne sont pas détectés, mais parfois un nystagmus et des symptômes cérébelleux légers sont possibles.
Le substrat morphologique de la maladie est reconnu comme un processus atrophique principalement dans la partie antérieure du ver cérébelleux. La maladie a été décrite pour la première fois en 1946 par M. Parker. Il se transmet sur le mode autosomique dominant. En 1987, avec l'ataxie paroxystique familiale, une diminution de l'activité de la pyruvate déshydrogénase des leucocytes sanguins à 50-60% du niveau normal a été trouvée. En 1977 R. Lafrance et al. a attiré l'attention sur l'effet prophylactique élevé du diacarbe, plus tard la flunarizine a été proposée pour le traitement de l'ataxie paroxystique familiale.
Ataxie cérébelleuse aiguë ou syndrome de Leyde-Westphal, est un complexe symptomatique bien défini, qui est une complication para-infectieuse. Elle survient plus souvent chez les enfants 1 à 2 semaines après une infection générale (grippe, typhus, salmonellose, etc.). Caractérisé par une ataxie statique et dynamique grossière, un tremblement intentionnel, une hypermétrie, une asynergie, un nystagmus, un discours chanté, une diminution du tonus musculaire. Dans le liquide céphalo-rachidien, une pléocytose lymphocytaire, une augmentation modérée des protéines, est détectée. Au début de la maladie, des vertiges, des troubles de la conscience, des convulsions sont possibles. Au scanner et à l'IRM, la pathologie n'est pas détectée. Le cours est bénin. Dans la plupart des cas, après quelques semaines ou mois - récupération complète, parfois - troubles résiduels sous forme d'insuffisance cérébelleuse légère.
Maladie de Marie-Foix-Alajuanin - atrophie corticale symétrique tardive du cervelet avec une lésion prédominante des neurones piriformes (cellules de Purkinje) et de la couche granuleuse du cortex, ainsi que de la partie buccale du vermis cérébelleux et dégénérescence olive. Elle se manifeste chez les personnes de 40 à 75 ans présentant des troubles de l'équilibre, de l'ataxie, des troubles de la marche, des troubles de la coordination et une diminution du tonus musculaire, principalement au niveau des jambes ; le tremblement intentionnel dans les mains n'est pas très prononcé. Les troubles de la parole sont possibles, mais ne font pas partie des signes obligatoires de la maladie. La maladie a été décrite en 1922 par les neuropathologistes français P. Marie, Ch. Foix et Th. Alajouanine. La maladie est sporadique. L'étiologie de la maladie n'a pas été élucidée. Il existe des opinions sur le rôle provocateur de l'intoxication, principalement l'abus d'alcool, ainsi que l'hypoxie, fardeau héréditaire. Le tableau clinique est confirmé par les données CT de la tête, qui révèlent une diminution prononcée du volume du cervelet dans le contexte de processus atrophiques diffus dans le cerveau. De plus, un niveau élevé d'aminotransférases dans le plasma sanguin est reconnu comme caractéristique (Ponomareva E.N. et al., 1997).
