7.1. СТРУКТУРА, ВРСКИ И ФУНКЦИИ НА МОЗОК
Малиот мозок (церебелум) се наоѓа под дупликат дура матер познат како преглед на малиот мозок(tentorium cerebelli), кој ја дели черепната празнина на два нееднакви простори - супатенторијален и субтенторијален. В субтенторијален простор,чие дно е задната кранијална јама, покрај малиот мозок, е и мозочното стебло. Волуменот на малиот мозок во просек е 162 cm 3. Неговата тежина варира помеѓу 136-169 g.
Малиот мозок се наоѓа над мостот и наддолжната медула. Заедно со горните и долните церебрални едра, тој го сочинува покривот на четвртата комора на мозокот, чие дно е таканаречената ромбоидна фоса (види Поглавје 9). Над малиот мозок се наоѓаат окципиталните лобуси на големиот мозок, одвоени од него со тенториумот на малиот мозок.
Во малиот мозок, има две хемисфери(hemispherum cerebelli). Помеѓу нив, во сагитталната рамнина над IV комора на мозокот, се наоѓа филогенетски најстариот дел од малиот мозок - неговиот црв(vermis cerebelli). Вермисот и церебеларните хемисфери се фрагментирани во лобули со длабоки попречни жлебови.
Малиот мозок е составен од сива и бела материја. Сивата материја го формира церебеларниот кортекс и спарените јадра церебелови јадра лоцирани во нејзината длабочина (сл. 7.1). Најголемите од нив се нерамни јадра(nucleus dentatus) - се наоѓа во хемисферите. Во централниот дел на црвот има јадра на шатор(јадра
Ориз. 7.1.Церебеларни јадра.
1 - заби јадро; 2 - плутано јадро; 3 - јадрото на шаторот; 4 - сферично јадро.
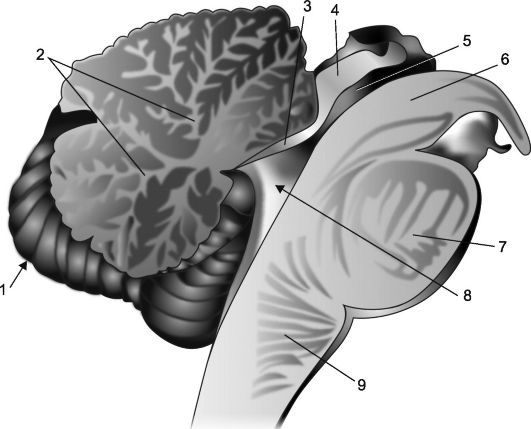
Ориз. 7.2.Сагитален дел на малиот мозок и мозочното стебло.
1 - малиот мозок; 2 - "дрво на животот"; 3 - преден мозок едро; 4 - плоча на четворката; 5 - аквадукт на мозокот; 6 - ногата на мозокот; 7 - мост; 8 - IV комора, нејзиниот хориоиден плексус и шатор; 9 - продолжена медула.
fastigii), меѓу нив и назабените јадра се сферичнии плутани јадра(nuctei.globosus et emboliformis).
Поради фактот што кортексот ја покрива целата површина на малиот мозок и продира во длабочините на неговите бразди, на сагитален дел од малиот мозок, неговото ткиво има шема на лист, чии вени се формирани од бела материја (сл. 7.2), што го сочинува т.н дрво на животот на малиот мозок (arbor vitae cerebelli). Во основата на дрвото на животот има засек во облик на клин, кој е горниот дел од шуплината на IV комора; рабовите на оваа вдлабнатина го формираат неговиот шатор. Покривот на шаторот е церебеларниот црв, а неговите предни и задни ѕидови се тенки церебрални плочи познати како предни и задни мозокот едра(vella medullare anterior et posterior).
Некои информации за церебеларна архитектоника,давајќи основа да се процени функцијата на неговите компоненти. Имаат церебеларниот кортексПостојат два клеточни слоеви: внатрешниот е грануларен, кој се состои од мали зрнести клетки, а надворешниот е молекуларен. Помеѓу нив има голем број големи клетки во облик на круша кои го носат името на чешкиот научник И. Пуркиње кој ги опишал (Пуркиње И., 1787-1869).
Импулсите влегуваат во церебеларниот кортекс преку мовни и ползечки влакна кои продираат во него од белата материја, кои ги сочинуваат аферентните патишта на малиот мозок. Преку мовливи влакна, импулси од 'рбетниот мозок
вестибуларните јадра и јадрата на понсот се пренесуваат во клетките на зрнестиот слој на кортексот. Аксоните на овие клетки, заедно со ползечките влакна кои минуваат низ зрнестиот слој во транзит и носат импулси од долните маслинки до малиот мозок, стигнуваат до површинскиот, молекуларен слој на малиот мозок. Овде, аксоните на клетките на зрнестиот слој и ползечките влакна се делат во форма на Т, а во молекуларниот слој нивните гранки заземаат насока надолжна до површината на малиот мозок. Импулсите што стигнале до молекуларниот слој на кортексот, поминувајќи низ синаптичките контакти, паѓаат на разгранетите дендрити на клетките на Пуркиние кои се наоѓаат овде. Потоа тие ги следат дендритите на Пуркиниевите клетки до нивните тела, лоцирани на границата на молекуларните и зрнестите слоеви. Потоа, по аксоните на истите клетки кои го преминуваат зрнестиот слој, тие продираат во длабочината на белата маса. Аксоните на Пуркиниевите клетки завршуваат во јадрата на малиот мозок. Главно во назабеното јадро. Еферентните импулси кои доаѓаат од малиот мозок долж аксоните на клетките што го сочинуваат неговото јадро и учествуваат во формирањето на церебеларните педуни го напуштаат малиот мозок.
Малиот мозок има три пара нозе:дното, средината и врвот. Долниот дел на ногата го поврзува со продолжениот мозок, средината - со мостот, горниот дел - со средниот мозок. Нозете на мозокот ги сочинуваат патиштата кои носат импулси до и од малиот мозок.
Церебеларниот вермис обезбедува стабилизирање на центарот на гравитација на телото, негова рамнотежа, стабилност, регулирање на тонот на реципрочните мускулни групи, главно вратот и трупот и појавата на физиолошки церебеларни синергии кои ја стабилизираат рамнотежата на телото.
За успешно одржување на рамнотежата на телото, малиот мозок постојано добива информации што минуваат по спиноцеребеларните патишта од проприоцепторите на различни делови од телото, како и од вестибуларните јадра, долните маслинки, ретикуларната формација и други формации вклучени во контролирањето на положба на делови од телото во просторот. Повеќето од аферентните патишта кои водат до малиот мозок минуваат низ долниот церебеларен педикул, некои од нив се наоѓаат во горниот церебеларен педикул.
Импулси на проприоцептивна чувствителност, одење до малиот мозок, како и другите сензорни импулси, следејќи ги дендритите на првите сензорни неврони, стигнуваат до нивните тела лоцирани во 'рбетните јазли. Последователно, импулсите што одат до малиот мозок долж аксоните на истите неврони се насочени кон телата на вторите неврони, кои се наоѓаат во внатрешните делови на основата на задните рогови, формирајќи т.н. Кларковите столбови. Нивните аксони паѓаат во страничните делови на страничните жици на 'рбетниот мозок, каде што се формираат спиноцеребеларни патишта, во овој случај, дел од аксоните паѓа во страничниот столб од истата страна и се формира таму задниот спиноцеребеларен тракт на Flexig (tractus spinocerebelaris posterior). Друг дел од аксоните на клетките на задните рогови поминува на другата страна на 'рбетниот мозок и навлегува во спротивниот страничен мозок, формирајќи се во него преден спиноцеребеларен тракт на Говерс (tractus spinocerebelaris anterior). Спиноцеребеларните патишта, зголемувајќи се во волуменот на ниво на секој сегмент на 'рбетниот столб, се издигнуваат до продолжетокот на медулата.
Во продолжениот мозок, задниот спиноцеребеларен пат отстапува во страничната насока и, откако ќе помине низ долниот церебеларен педикул, продира во малиот мозок. Предниот спиноцеребеларен пат минува низ продолжениот мозок, мозочниот мозок, и стигнува до средниот мозок, на чие ниво го прави својот втор пресек во предниот церебрален превез и поминува во малиот мозок преку горниот церебеларен педункул.
Така, од двата 'рбетни патишта, едниот никогаш не е вкрстен (непрекрстена патека на Флексиг), а другиот двапати поминува на спротивната страна (двапати преминал од Гуверс). Како резултат на тоа, и двајцата спроведуваат импулси од секоја половина од телото, главно до хомолатералната половина на малиот мозок.
Покрај спиноцеребеларните патишта на Флексиг, импулсите кон малиот мозок минуваат низ долниот церебеларен педикул по вестибулоцеребеларен тракт (tractus vestibulocerebellaris), почнувајќи главно во горното вестибуларно јадро на анкилозантен спондилитис и по оливомоцеребеларен тракт (tractus olivocerebelaris) кој доаѓа од долната маслинка. Дел од аксоните на клетките на тенките и клиновидни јадра, не учествува во формирањето на булботаламичниот тракт, во форма на надворешни лачни влакна (fiber arcuatae externae) исто така навлегува во малиот мозок преку долниот церебеларен педункул.
Преку средните нозе, малиот мозок прима импулси од церебралниот кортекс. Овие импулси минуваат низ кортикално-церебелопонтински патишта, кои се состојат од два неврони. Телата на првите неврони се наоѓаат во церебралниот кортекс, главно во кората на задните делови на фронталните лобуси. Нивните аксони минуваат како дел од сјајната круна, предната нога на внатрешната капсула и завршуваат во јадрата на мостот. Аксони на клетки на втори неврони, чии тела се наоѓаат во нивните јадра на мостот, одете на нејзината спротивна страна и направете, по пресекот, средната церебеларна педикула,
завршувајќи на спротивната хемисфера на малиот мозок.
Некои од импулсите што се појавија во церебралниот кортекс стигнуваат до спротивната хемисфера на малиот мозок, носејќи информации не за произведеното, туку само за планираното активно движење. Откако ги доби таквите информации, малиот мозок веднаш испраќа импулси кои ги коригираат доброволните движења, главно, со гаснење на инерција и најрационално регулирање на реципрочниот мускулен тонус - мускулни агонисти и антагонисти. Како резултат на тоа, еден вид на еметрија,правејќи ги доброволните движења јасни, усовршени, лишени од несоодветни компоненти.
Патеките што го напуштаат малиот мозок се составени од аксоните на клетките, чии тела ги формираат неговите јадра. Повеќето еферентни патишта, вклучувајќи ги патеките од назабените јадра, оставете го малиот мозок низ горниот дел од ногата. На ниво на долните туберкули на четворката, еферентниот церебеларен тракт се вкрстува (пресек на горните церебеларни нозе на Вернекинг). По преминувањето на секој од нив допира до црвените јадра на спротивната страна на средниот мозок. Во црвените јадра, церебеларните импулси се префрлаат на следниот неврон, а потоа се движат по аксоните на клетките, чии тела се вградени во црвените јадра. Овие аксони се формираат во црвено-рбетните патишта (tracti rubro spinalis), патеките на Монаков, кои набргу потоа излезите од црвените јадра се подложени на вкрстување (крст за гуми или крст на пастрмка), по што се спуштаат во 'рбетниот мозок. Во 'рбетниот мозок, црвено-нуклеарните' рбетни патишта се наоѓаат во страничните жици; нивните составни влакна завршуваат на клетките на предните рогови на 'рбетниот мозок.
Целата еферентна патека од малиот мозок до клетките на предните рогови на 'рбетниот мозок може да се нарече церебеларно-црвено-нуклеарно-рбетниот (tractus cerebello-rubrospinalis). Два пати нафрлува (пресек на горните церебеларни педуни и пресек на оперкулумот) и на крајот ја поврзува секоја церебеларна хемисфера со периферните моторни неврони лоцирани во предните рогови на хомолатералната половина на 'рбетниот мозок.
Од јадрата на церебеларниот вермис, еферентните патишта главно одат низ долниот церебеларен педикул до ретикуларната формација на мозочното стебло и вестибуларните јадра. Оттука, по должината на ретикулоспиналните и вестибулоспиналните патишта кои минуваат по предните жици на 'рбетниот мозок, тие исто така стигнуваат до клетките на предните рогови. Дел од импулсите што доаѓаат од малиот мозок, минувајќи низ вестибуларните јадра, влегуваат во медијалниот надолжен сноп, стигнуваат до јадрата III, IV и VI на кранијалните нерви кои обезбедуваат движење на очните јаболка и влијае на нивната функција.
Накратко, треба да се нагласи следново:
1. Секоја половина од малиот мозок прима импулси главно а) од хомолатералната половина на телото, б) од спротивната хемисфера на мозокот, која има кортико-спинални врски со истата половина од телото.
(2) Од секоја половина на малиот мозок, еферентните импулси се насочени кон клетките на предните рогови на хомолатералната половина на 'рбетниот мозок и до јадрата на кранијалните нерви кои обезбедуваат движење на очните јаболка.
Оваа природа на церебеларните врски овозможува да се разбере зошто, кога е зафатена една половина од малиот мозок, церебеларните нарушувања се јавуваат главно во истиот, т.е. хомолатерална, половина од телото. Ова е особено изразено кога се зафатени церебеларните хемисфери.
7.2. ИСТРАЖУВАЊЕ НА ФУНКЦИИТЕ НА МОЛИЦИОТ
И КЛИНИЧКИ МАНИФЕСТАЦИИ НА НЕГОВИТЕ ПОРАЗИ
Со оштетување на малиот мозок, карактеристични се нарушувања на статиката и координацијата на движењата, мускулна хипотонија и нистагмус.
Церебеларна лезија најпрво неговиот црв,доведува до нарушување на статиката - способност да се одржи стабилна позиција на центарот на гравитација на човечкото тело, рамнотежа, стабилност. Кога оваа функција е нарушена, статична атаксија (од грчката атаксија - нарушување, нестабилност). Забележана е нестабилност на пациентот. Затоа, во стоечка положба, широко ги раширува нозете, балансира со рацете. Особено јасно статична атаксија е откриена со вештачко намалување, особено во областа на поддршката во позата Ромберг. Пациентот е поканет да застане, цврсто да ги движи стапалата и малку да ја подигне главата. Во присуство на церебеларни нарушувања, се забележува нестабилност на пациентот во оваа положба, неговото тело се ниша, понекогаш се „влече“ во одредена насока, а ако пациентот не е поддржан, може да падне. Во случај на оштетување на церебеларниот црв, пациентот обично се ниша од страна на страна и често паѓа назад. Со патологија на церебеларната хемисфера, постои тенденција да падне главно кон патолошкиот фокус. Ако статичкото нарушување е умерено изразено, полесно е да се идентификува во т.н комплицираноили сензибилизирана поза на Ромберг. Од пациентот се бара да ги стави стапалата во една линија така што палецот на едната нога да лежи на петицата на другата. Проценката на стабилноста е иста како и во вообичаената позиција на Ромберг.
Нормално, кога човек стои, мускулите на нозете му се напнати. (реакција на поддршка), со закана да падне на страна, неговата нога од оваа страна се движи во иста насока, а другата нога се симнува од подот (реакција на скок). Со оштетување на малиот мозок (главно црвот), реакциите на пациентот се нарушени
поддршка и скок. Повреда на реакцијата на поддршка се манифестира со нестабилност на пациентот во стоечката положба, особено во положбата Ромберг. Прекршувањето на реакцијата на скок води до фактот дека ако лекарот, стоејќи зад пациентот и осигурувајќи го, го турка пациентот во една или друга насока, тогаш пациентот паѓа со мало туркање (симптом на туркање).
Со оштетување на малиот мозок, одењето на пациентот обично се менува поради развојот статолокомоторна атаксија. Церебеларна одење на многу начини наликува на одењето на пијана личност, затоа понекогаш се нарекува „одење на пијаница“. Поради нестабилност, пациентот оди несигурно, раширувајќи ги нозете широко, додека е „фрлен“ од страна на страна. И кога церебеларната хемисфера е оштетена, таа отстапува при одење од дадена насока кон патолошкиот фокус. Нестабилноста е особено изразена при свиоци. Ако атаксијата е изразена, тогаш пациентите целосно ја губат способноста да го контролираат своето тело и не само што можат да стојат и да одат, туку дури и да седат.
Доминантната лезија на церебеларните хемисфери доведува до нарушување на неговите антиинертни ефекти, особено до појава на кинетичка атаксија. Се манифестира со незгодност на движењата и особено е изразен со движења кои бараат прецизност. За откривање на кинетичка атаксија, се прават тестови за координација на движењата. Некои од нив се опишани подолу.
Тест за дијадохокинеза (од грчки. diadochos - низа). Пациентот е поканет да ги затвори очите, да ги истегне рацете напред и брзо, ритмички да леже и да навлезе во рацете. Во случај на оштетување на церебеларната хемисфера, движењата на раката на страната на патолошкиот процес излегуваат пообемни (последица на дисметрија, поточно хиперметрија), како резултат на тоа, раката почнува да заостанува. Ова укажува на присуство на адиадохокинеза.
Тест со прст. Пациент со затворени очи треба да ја повлече раката, а потоа, полека, со показалецот да го допре врвот на носот. Во случај на церебеларна патологија, раката на страната на патолошкиот фокус прави прекумерно движење во волуменот (хиперметрија),како резултат на што пациентот промаши. Тестот со прст-нос открива карактеристика на церебеларна патологија церебеларен (намерен) тремор, чија амплитуда се зголемува како што прстот се приближува до целта. Овој тест ви овозможува да ја идентификувате таканаречената брадителекинезија. (симптом на узда):недалеку од целта, движењето на прстот се забавува, понекогаш дури и паузира, а потоа продолжува повторно.
Тест со прст-прст. Пациент со затворени очи е поканет да ги рашири рацете широко, а потоа да ги приближи показалците, обидувајќи се да го внесе прстот во прстот, додека, како и кај тестот со прст, се открива намерен тремор и симптом на уздата.
Калканеален тест на коленото (сл. 7.3). Од пациентот, кој лежи на грб со затворени очи, се бара да ја крене едната нога високо, а потоа со петата да го удри коленото од другата нога. Со патологија на малиот мозок, пациентот не може или му е тешко да ја внесе петата во коленото на другата нога, особено кога прави тест со ногата хомолатерална на погодената церебеларна хемисфера. Ако, сепак, петицата стигне до коленото, тогаш се предлага да се држи, малку допирајќи ја предната површина на долниот дел на ногата, до зглобот на глуждот, додека во случај на патологија на церебеларна, петата се лизга од долниот дел на ногата. времето во една или друга насока.

Ориз. 7.3.Калканеален тест на коленото.
Индикативен тест: Пациентот е повикан неколку пати со показалецот да го удри гумениот врв на чеканот, кој е во раката на испитувачот. Во случај на церебеларна патологија во раката на пациентот на страната на погодената церебеларна хемисфера, постои неусогласеност поради дисметрија.
Симптом на Том-Џументи: Ако пациентот зема предмет, како што е чашата, тој прекумерно ги шири прстите.
Церебеларен нистагмус. Грчењето на очното јаболко при гледање на страните (хоризонтален нистагмус) се смета како последица на намерно тремор на очните јаболка (види поглавје 30).
Нарушување на говорот: Говорот ја губи својата флуидност, станува експлозивен, фрагментиран, опеан како церебеларна дизартрија (види Поглавје 25).
Промена на ракопис: Поради нарушување во координацијата на движењата на рацете, ракописот станува нерамномерен, буквите се деформирани, претерано големи (мегалографија).
Пронаторен феномен: Од пациентот се бара да ги држи рацете испружени во положба на лежење, додека спонтана пронација набргу се јавува на страната на погодената церебеларна хемисфера.
Симптом на Хоф-Шилдер: Ако пациентот ги држи рацете испружени напред, тогаш на страната на погодената хемисфера, раката наскоро се повлекува нанадвор.
Феномен на имитација. Пациентот со затворени очи треба брзо да и даде на раката положба слична на онаа што испитувачот претходно и ја дал на другата рака. Кога церебеларната хемисфера е оштетена, хомолатералната рака прави движење што е прекумерно во амплитуда.
Феноменот на Доиников. Феномен на прст. Пациентот што седи е поканет да ги стави лежечките раце со раздвоени прсти на бутовите и да ги затвори очите. Во случај на лезија на малиот мозок на страната на патолошкиот фокус, наскоро се јавува спонтана флексија на прстите и пронација на раката и подлактицата.
Симптом на Стјуарт-Холмс. Испитувачот бара од пациентот кој седи на столот да ги свитка лежечките подлактици и во исто време, земајќи ги рацете за зглобовите, му дава отпор. Ако во исто време одеднаш ги ослободите рацете на пациентот, тогаш раката на засегнатата страна, свиткувајќи се по инерција, силно ќе го удри во градите.
Мускулна хипотензија. Поразот на церебеларниот вермис обично води до дифузна мускулна хипотензија. Со поразот на церебеларната хемисфера, пасивните движења откриваат намалување на мускулниот тонус на страната на патолошкиот процес. Мускулната хипотонија доведува до можност за хиперекстензија на подлактицата и потколеницата (симптом на Олшански) со пасивни движења, до изгледот симптоми на висечка рака или нога со нивното пасивно тресење.
Патолошки церебеларни асинергии. Прекршувањата на физиолошките синергии при сложени моторни дејства се откриваат, особено, за време на следните тестови (сл. 7.4).
1. Асинергија според Бабински во стоечка положба.Ако пациентот кој стои со поместени нозе се обиде да се наведне наназад, фрлајќи ја главата назад, тогаш нормално во овој случај се јавува флексија на коленото зглобовите. Кај церебеларната патологија поради асинергија, ова пријателско движење е отсутно, а пациентот, губејќи рамнотежа, паѓа назад.
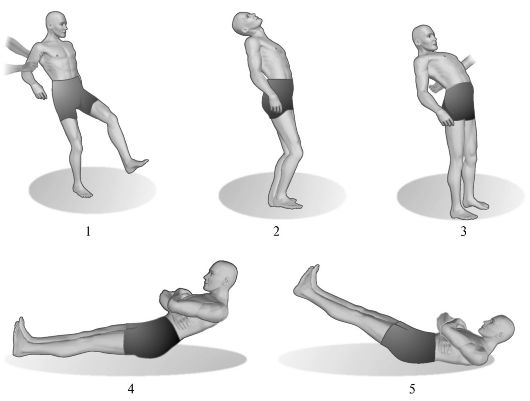
Ориз. 7.4.Церебеларна асинергија.
1 - одење на пациент со тешка церебеларна атаксија; 2 - навалувањето на телото на грбот е нормално; 3 - со оштетување на малиот мозок, пациентот, свиткувајќи се наназад, не може да одржува рамнотежа; 4 - вршење тест за церебеларна асинергија според Бабински од здраво лице; 5 - извршување на истиот тест кај пациенти со церебеларни лезии.
2. Асинергија според Бабински во лежечка положба.Пациентот, легнат на цврста рамнина со испружени нозе, раширени до ширината на рамениот појас, е поканет да ги прекрсти рацете преку градите и потоа да седне. Во присуство на церебеларна патологија поради отсуство на пријателска контракција на глутеалните мускули (манифестација на асинергија), пациентот не може да ги фиксира нозете и карлицата на потпорната област, како резултат на тоа, нозете се креваат и не може да седне. Значењето на овој симптом не треба да се прецени кај постари пациенти, кај луѓе со млитав или дебел абдоминален ѕид.
Сумирајќи го горенаведеното, треба да се нагласи различноста и важноста на функциите што ги извршува малиот мозок. Како дел од сложениот регулаторен механизам за повратни информации, малиот мозок делува како фокусна точка за балансирање на телото и одржување на мускулниот тонус. Како што забележува P. Duus (1995), малиот мозок обезбедува способност да врши дискретни и прецизни движења,авторот разумно верува дека малиот мозок работи како компјутер, следејќи ги и координирајќи сетилни информации на влезот и симулирајќи моторни сигнали на излезот.
7.3. МУЛТИСИСТЕМСКИ ДЕГЕНЕРАЦИИ
Со знаци на церебеларна патологија
Мултисистемските дегенерации се група на невродегенеративни болести, чија заедничка карактеристика е мултифокалната природа на лезијата со вклучување на различни функционални и невротрансмитерски системи на мозокот во патолошкиот процес и, според тоа, полисистемската природа на клиничките манифестации.
7.3.1. Церебеларна атаксија
Спиноцеребеларните атаксии вклучуваат прогресивни наследни дегенеративни заболувања, во кои главно се засегнати структурите на малиот мозок, мозочното стебло и патиштата на 'рбетниот мозок, кои главно се поврзани со екстрапирамидалниот систем.
7.3.1.1. Наследна атаксија на Фридрајх
Наследна болест опишана во 1861 година од германскиот невропатолог N. Friedreich (Friedreich N., 1825-1882). Се наследува на автосомно рецесивен начин или (поретко) на автосомно доминантен начин со нецелосна пенетрација и променлива генска експресија. Можни се и спорадични случаи на болеста.
Патогенезаболест не е наведена. Конкретно, нема идеја за примарниот биохемиски дефект што ја сочинува неговата основа.
Патоморфологија.Патолошките студии откриваат изразено разредување на 'рбетниот мозок поради атрофични процеси во неговите задни и странични жици. Како по правило, страдаат клиновидните (Burdach) и нежните (Gaulle) патишта и спиноцеребеларните патишта на Govers и Fleksig, како и вкрстената пирамидална патека која содржи
многу влакна поврзани со екстрапирамидалниот систем. Дегенеративните процеси се изразени и во малиот мозок, во неговата бела маса и во нуклеарниот апарат.
Клинички манифестации. Болеста се манифестира кај деца или млади помлади од 25 години. С.Н. Давиденков (1880-1961) забележа дека почесто клиничките знаци на болеста се јавуваат кај деца на возраст од 6-10 години. Првиот знак на болеста е обично атаксија. Пациентите доживуваат несигурност, тетеравење при одење, промени во одењето (при одење, нозете се широко разделени). Одењето кај Фридрајховата болест може да се нарече табетичко-церебеларна, бидејќи неговите промени се предизвикани од комбинација на чувствителна и церебеларна атаксија, како и обично изразено намалување на мускулниот тонус. Карактеристични се и нарушувања на статиката, дискоординација во рацете, намерен тремор, дизартрија. Можен нистагмус, губење на слухот, елементи на пеење говор, знаци на пирамидална инсуфициенција (хиперрефлексија на тетивите, патолошки рефлекси на стапалата, понекогаш благо зголемување на мускулниот тонус), императивен нагон за мокрење, намалена сексуална потенција. Понекогаш се појавува атетоидна хиперкинеза.
Раното пореметување на длабоката чувствителност доведува до прогресивно намалување на тетивните рефлекси: прво на нозете, а потоа и на рацете. Со текот на времето, се формира хипотрофија на мускулите на дисталните нозе. Карактеристично е присуството на аномалии во развојот на скелетот. Прво на сите, ова се манифестира со присуство Нозете на Фридрајх: стапалото е скратено, „шупливо“, со многу висок лак. Главните фаланги на нејзините прсти не се свиткани, а останатите се свиткани (сл. 7.5). Можна деформација на 'рбетот, градите. Често има манифестации на кардиопатија. Болеста напредува бавно, но стабилно доведува до инвалидитет на пациентите кои на крајот стануваат приковани за кревет.
Третман. Патогенетски третман не е развиен. Препишете лекови кои го подобруваат метаболизмот во структурите на нервниот систем, агенси за зајакнување. Со тежок деформитет на стапалата, индицирани се ортопедски чевли.
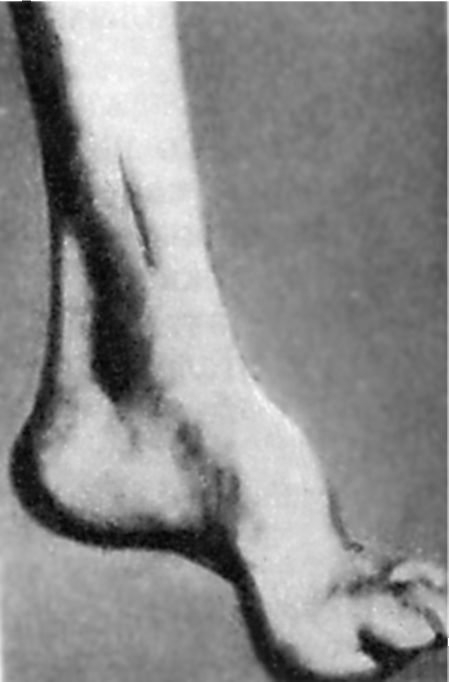
Ориз. 7.5.Ногата на Фридрајх.
7.3.1.2. Наследна церебеларна атаксија (Пјер Мари-ова болест)
Станува збор за хронично прогресивно наследно заболување, кое се манифестира на возраст од 30-45 години, со бавно растечки церебеларни нарушувања во комбинација со знаци на пирамидална инсуфициенција, а карактеристични се статична и динамична церебеларна атаксија, намерни тремори, скандиран говор, хиперрефлексија на тетивите. Можни клонуси, патолошки пирамидални рефлекси, страбизам, намален вид, стеснување на видните полиња поради примарна атрофија на оптичките нерви и пигментна дегенерација на мрежницата. Текот на болеста полека напредува. Постои намалување на големината на малиот мозок, дегенерација на клетките
Пуркиње, инфериорни маслинки, спиноцеребеларни патишта. Се наследува на автосомно доминантен начин. Болеста беше опишана во 1893 година од францускиот невропатолог R. Marie (1853-1940).
Во моментов, не постои едногласност во разбирањето на терминот „болест на Пјер Мари“, а дискутабилно е прашањето за можноста за нејзино раздвојување во независна нозолошка форма.
Не е развиен третман. Обично, се користат метаболички активни и ресторативни, како и симптоматски агенси.
7.3.2. Оливиопонтоцеребеларна дистрофија (Дежерин-Том болест)
Ова е група на хронични прогресивни наследни болести, во кои дистрофичните промени се развиваат главно во малиот мозок, долните маслинки, во сопствените јадра на понсот и во мозочните структури поврзани со нив.
Со развојот на болеста на млада возраст, околу половина од случаите се наследуваат на доминантен или рецесивен начин, а останатите се спорадични. Во спорадични случаи на болеста, почести се манифестациите на акинетичко-ригиден синдром и прогресивна автономна инсуфициенција. Просечната возраст на пациентот со манифестација на наследна форма на болеста во фенотипот е 28 години, со спорадична - 49 години, просечниот животен век е 14,9 и 6,3 години, соодветно. Во спорадичната форма, покрај атрофијата на маслинките, понсот и малиот мозок, почесто се среќаваат лезии на страничните жици на 'рбетниот мозок, супстанција нигра и стриатум, синкаста дамка во ромбоидната јама на четвртата комора на мозокот. .
Карактеристични се симптомите на растечкиот церебеларен синдром. Можни нарушувања на чувствителноста, елементи на булбарни и акинетичко-ригидни синдроми, хиперкинеза, особено миоритмии на јазикот и мекото непце, офталмопареза, намалена визуелна острина, интелектуални нарушувања. Болеста била опишана во 1900 година од француските невропатолози Ј. Дежерин и А. Томас.
Болеста често дебитира со пореметувања во одењето - можна е нестабилност, дискоординација, неочекувани падови. Овие нарушувања може да бидат единствената манифестација на болеста во рок од 1-2 години. Во иднина, се појавуваат и растат нарушувања на координацијата во рацете: манипулациите со мали предмети се тешки, ракописот е нарушен, се јавува намерен тремор. Говорот станува повремен, заматен, со назална нијанса и ритам на дишење што не одговара на структурата на говорот (пациентот зборува како да го задавуваат). Во оваа фаза на болеста, се приклучуваат манифестации на прогресивна автономна инсуфициенција, се појавуваат знаци на акинетичко-ригиден синдром. Понекогаш доминантни симптоми за пациентот се дисфагија, напади на ноќно гушење. Тие се развиваат во врска со мешана пареза на булбарните мускули и можат да бидат опасни по живот.
Во 1970 година, германските невропатолози Б.В. Конигсмарк и Л.П. Вајнер издвои 5 главни типовиоливопонтоцеребеларна дистрофија, која се разликува или во клинички и морфолошки манифестации, или во вид на наследување.
Јас тип (тип Менцел). На возраст од 14-70 (почесто 30-40) години се манифестира атаксија, дизартрија, дисфонија, мускулна хипотонија, во доцна фаза - груб тремор на главата, трупот, рацете, мускулите, знаци на акинетика- ригиден синдром. Можни патолошки пирамидални знаци, пареза на погледот, надворешна и внатрешна офталмоплегија, нарушувања на чувствителноста, деменција. Се наследува на автосомно доминантен начин. Како самостојна форма е издвоена во 1891 година од П. Менцел.
II тип (тип Фиклер-Винклер) ... На возраст од 20-80 години се манифестира атаксија, намален мускулен тонус и тетивни рефлекси. Се наследува на автосомно рецесивен начин. Можни се спорадични случаи.
III тип со ретинална дегенерација. Се манифестира во детска или млада (до 35 години) возраст атаксија, тремор на главата и екстремитетите, дизартрија, знаци на пирамидална инсуфициенција, прогресивно намалување на видот со исход во слепило; можен нистагмус, офталмоплегија, понекогаш дисоцирани сензорни нарушувања. Се наследува на автосомно доминантен начин.
IV тип (тип Jester-Highmaker). На возраст од 17-30 години, тој дебитира со церебеларна атаксија или знаци на долна спастична парапареза, во двата случаи, веќе во рана фаза на болеста, се формира комбинација од овие манифестации, на кои елементи на булбарен синдром, пареза на мускулите на лицето, а потоа се додаваат и длабоки нарушувања на чувствителноста. Доминантна наследена.
В тип на. Се манифестира на возраст од 7-45 години, можна е атаксија, дизартрија, знаци на акинетичко-ригиден синдром и други екстрапирамидални нарушувања, прогресивна офталмоплегија и деменција. Доминантна наследена.
7.3.3. Дегенерација на оливоруброцеребеларна (Lejeune-Lermitte синдром, Лермитова болест)
Болеста се карактеризира со прогресивна атрофија на малиот мозок, главно на неговиот кортекс, назабените јадра и горните церебеларни педуни, долните маслинки и црвените јадра. Се манифестира првенствено со статична и динамична атаксија, во иднина можни се други знаци на церебеларен синдром и оштетување на мозочното стебло. Оваа болест е опишана од француските невропатолози J. Lhermitte (Lhermitte J.J., 1877-1959) и J. Lezhon (Lejonne J., роден во 1894 година).
7.3.4. Мултисистемска атрофија
Во последниве децении, спорадична, прогресивна невродегенеративна болест наречена мултисистемска атрофија е идентификувана како независна форма. Се карактеризира со комбинирана лезија на базалните ганглии, малиот мозок, мозочното стебло, 'рбетниот мозок. Главните клинички манифестации: паркинсонизам, церебеларна атаксија, знаци на пирамидална и автономна инсуфициенција (Левин О.С., 2002). Во зависност од доминацијата на одредени карактеристики на клиничката слика, се разликуваат три типа на мултисистемска атрофија.
1) оливопонтоцеребеларен тип, кој се карактеризира со доминација на знаци на церебеларен напад;
2) стрионигрален тип, кај кој доминираат знаци на паркинсонизам;
3) синдром Шаи-Дрејџер, кој се карактеризира со доминација во клиничката слика на знаци на прогресивна автономна инсуфициенција со симптоми на ортостатска артериска хипотензија.
Основата на мултисистемската атрофија е селективна дегенерација на одредени области на претежно сивата материја на мозокот со оштетување на невроните и глијалните елементи. Причините за дегенеративни манифестации во мозочното ткиво и денес остануваат непознати. Манифестациите на мултисистемска атрофија на оливопонтоцеребеларниот тип се поврзани со оштетување на Пуркиниевите клетки во церебеларниот кортекс, како и на невроните на долните маслинки, понтоцеребеларните јадра, демиелинизација и дегенерација, главно на понтоцеребеларните патишта.
Церебеларните нарушувања обично се статична и динамична атаксија со нарушено локомоторно движење. Се карактеризира со нестабилност во положбата Ромберг, атаксија при одење, дисметрија, адиадохокинеза, намерен тремор, може да има нистагмус (хоризонтална вертикална, тепање надолу), наизменично и бавно следење на движењата на погледот, нарушена конвергенција на очите, пеан говор.
Мултисистемската атрофија обично се јавува во зрелоста и брзо напредува. Дијагнозата се базира на клинички докази и се карактеризира со комбинација на знаци на паркинсонизам, церебеларна инсуфициенција и автономни нарушувања. Третманот на болеста не е развиен. Времетраењето на болеста - во рок од 10 години, завршува со смрт.
7.4. ДРУГИ БОЛЕСТИ ПОВРЗАНИ СО СИМПТОМИ НА ЦЕРЕБРАЛНИ БОЛЕСТИ
Ако пациентот покажува знаци на церебеларна лезија, тогаш во повеќето случаи, пред сè треба да размислите за можностацеребеларни тумори(астроцитом, ангиобластом, медулобластом, метастатски тумори) или мултиплекс склероза. На церебеларни туморисе појавуваат рани знаци на интракранијална хипертензија. Кај мултиплекс склероза, обично е можно да се идентификуваат, покрај церебеларната патологија, клинички манифестации на оштетување на други структури на централниот нервен систем, првенствено визуелниот и пирамидалниот систем. Во класичната неврологија, карактеристиката на мултиплекс склерозаТријадата на Шарко: нистагмус, намерен тремор и скандиран говор и Нонеов синдром:нарушување на координацијата на движењата, дисметрија, скандиран говор и церебеларна асинергија.
Церебеларните нарушувања се големи и во посттрауматски Ман синдром,која се карактеризира со атаксија, дискоординација, асинергија, нистагмус. Траума или инфекција може да предизвикаат малиот мозок Голдштајн-Рајхман синдром:нарушувања на статиката и координацијата на движењата, асинергија, намерен тремор, намален мускулен тонус, хиперметрија, мегалографија, нарушена перцепција на масата (тежината) на предметот во рацете.
Нарушувањата на церебеларната функција, исто така, можат да бидат вродени по природа, манифестирајќи се, особено, Земановиот синдром:атаксија, одложен развој на говорот и последователно церебеларна дизартрија.
Вродена церебеларна атаксија Се манифестира со доцнење во развојот на моторните функции на детето (на возраст од 6 месеци не може да седи, почнува да оди доцна, додека одењето е атактично), како и со задоцнет говор, продолжено зачувување на дизартрија, понекогаш ментална ретардација, а микрокранијалните манифестации не се невообичаени. На КТ, церебеларните хемисфери се намалени. До околу 10-годишна возраст, обично се јавува компензација на мозочните функции, кои, сепак, може да се нарушат под влијание на штетни егзогени влијанија. Можни се и прогресивни форми на болеста.
Манифестација на вродена хипоплазија на малиот мозок е и Фанкони-Тарнеров синдром.Се карактеризира со нарушена статика и координација на движењата, нистагмус, кои обично се придружени со ментална ретардација.
Вродениот, исто така, вклучува автосомно рецесивен наследен тип, кој ретко се наоѓа Бетен-ова болест:Се карактеризира со вродена церебеларна атаксија, манифестирана во првата година од животот со нарушена статика и координација на движењата, нистагмус, нарушување на координацијата на погледот и умерена мускулна хипотонија. Можни се диспластични знаци. Детето доцни, понекогаш само на 2-3 години, почнува да ја држи главата, дури и подоцна - да стои, да оди, да зборува. Неговиот говор се менува според типот на церебеларна дизартрија. Можни се вегетативно-висцерални нарушувања, манифестации на имуносупресија. По неколку години, клиничката слика обично се стабилизира, пациентот до одреден степен се прилагодува на постоечките дефекти.
Спастична атаксија по предлог на A. Bell и E. Carmichel (1939), именувана е церебеларна атаксија наследена од автосомно доминантен тип, која се карактеризира со почеток на болеста на возраст од 3-4 години и се манифестира со комбинација на церебеларна атаксија со дизартрија, хиперрефлексија на тетивата и зголемен мускулен тонус според спастичен тип, додека можна е (но не задолжителни знаци на болеста) атрофија на оптичките нерви, дегенерација на мрежницата, нистагмус, окуломоторни нарушувања.
Автозомно доминантно се наследува Фелдман синдром(опишан од германскиот лекар Х. Фелдман, роден во 1919 г.): церебеларна атаксија, намерни тремори и рано побелување на косата. Се манифестира во втората деценија од животот и понатаму полека напредува, што доведува до инвалидитет по 20-30 години.
Доцна церебеларна атрофија или Том-ов синдромопишан во 1906 година од францускиот невролог А. Томас (1867-1963), обично се манифестира кај лица над 50 години со прогресивна атрофија на церебеларниот кортекс. Во фенотипот, се појавуваат знаци на церебеларен синдром, првенствено церебеларна статична и локомоторна атаксија, скандиран говор и промени во ракописот. Во далеку напредната фаза, можни се манифестации на пирамидална инсуфициенција.
Комбинацијата на церебеларни нарушувања со миоклонус се карактеризира со Лов миоклонична церебеларна дисинергија,или миоклонусна атаксија,со овој комплекс на симптоми во клиничката слика, се манифестира намерен тремор, миоклонус што се појавува во рацете, а подоцна добива генерализиран карактер, атаксија и дисинергија, нистагмус, скандиран говор, намален мускулен тонус. Тоа е последица на дегенерација на церебеларните јадра, црвените јадра и нивните врски, како и кортикално-субкортикалните структури.
Во напредна фаза на болеста, можни се епилептични напади и деменција. Прогнозата е лоша. Се однесува на ретка форма на прогресивна наследна атаксија. Се наследува на автосомно рецесивен начин. Обично се појавува на млада возраст. Нозолошката независност на комплексот на симптоми е оспорена. Американскиот невролог Р. Хант (1872-1937) ја опиша болеста во 1921 година.
Меѓу дегенеративните процеси, одредено место зазема Холмсова церебеларна дегенерација,или фамилијарна церебеларна атрофија,или прогресивна атрофија на церебеларниот систем, главно на назабените јадра, како и црвените јадра, додека манифестациите на демиелинизација се изразени во горниот церебеларен педикул. Се карактеризира со статична и динамична атаксија, асинергија, нистагмус, дизартрија, намален мускулен тонус, мускулна дистонија, тремор на главата, миоклонус. Епилептичните напади се појавуваат речиси истовремено. Интелигенцијата обично се чува. ЕЕГ покажува пароксизмална дисритмија. Болеста е препознаена како наследна, но типот на наследство не е наведен. Ја опишал болеста во 1907 година од англискиот невропатолог Г. Холмс
(1876-1965).
Алкохолна церебеларна дегенерација - последица на хронична алкохолна интоксикација. Главно е зафатен церебеларниот црв, при што примарно се манифестираат церебеларна атаксија и нарушена координација на движењата на нозете, додека движењата на рацете, окуломоторните и говорните функции се нарушени во многу помала мера. Обично оваа болест е придружена со изразено губење на меморијата во комбинација со полиневропатија.
се манифестира како церебеларна атаксија, која понекогаш може да биде единствениот клинички симптом поврзан со малигнен тумор, без локални знаци кои укажуваат на местото на неговото појавување. Паранеопластична церебеларна дегенерацијаможе да биде, особено, секундарна манифестација на рак на дојка или јајници.
Синдромот Баракер-Бордас-Руиз-Лара се манифестира како церебеларни нарушувања кои произлегуваат во врска со брзо прогресивна атрофија на малиот мозок. Синдром кај пациенти со бронхијален карцином придружен со општа интоксикација е опишан од современиот шпански лекар Л. Баракер-Бордас (роден во 1923 година).
Ретко се наоѓа рецесивна Х-хромозомска атаксија- наследна болест која се манифестира речиси само кај мажи со бавно прогресивна церебеларна инсуфициенција. Се пренесува во рецесивен, полово-поврзан тип.
Вреди да се забележи и фамилијарна пароксизмална атаксија,или периодична атаксија.Своето деби го прави почесто во детството, но може да се појави и подоцна - до 60 години. Клиничката слика е сведена на пароксизмални манифестации на нистагмус, дизартрија и атаксија, намален мускулен тонус, вртоглавица, гадење, повраќање, главоболка, кои траат од неколку минути до 4 недели.
Нападите на фамилијарна пароксизмална атаксија може да бидат поттикнати од емоционален стрес, физички замор, треска, внес на алкохол, додека помеѓу нападите фокални невролошки симптоми во повеќето случаи не се откриваат, но понекогаш се можни нистагмус и благи церебеларни симптоми.
Морфолошкиот супстрат на болеста е препознаен како атрофичен процес главно во предниот дел на церебеларниот црв. Болеста за прв пат беше опишана во 1946 година од М. Паркер. Се наследува на автосомно доминантен начин. Во 1987 година, со фамилијарна пароксизмална атаксија, беше откриено намалување на активноста на пируват дехидрогеназата на леукоцитите во крвта до 50-60% од нормалното ниво. Во 1977 година R. Lafrance et al. го привлече вниманието на високиот профилактички ефект на дијакарбот, подоцна флунаризин беше предложен за третман на фамилијарна пароксизмална атаксија.
Акутна церебеларна атаксија или Лајден-Вестфал синдром,е добро дефиниран комплекс на симптоми, кој е параинфективна компликација. Почесто се јавува кај деца 1-2 недели по општа инфекција (грип, тифус, салмонелоза итн.). Карактеризира со груба статичка и динамична атаксија, намерен тремор, хиперметрија, асинергија, нистагмус, скандиран говор, намален мускулен тонус. Во цереброспиналната течност, откриена е лимфоцитна плеоцитоза, умерено зголемување на протеините. На почетокот на болеста, можни се вртоглавица, нарушувања на свеста, конвулзии. На КТ и МРИ, патологијата не е откриена. Курсот е бениген. Во повеќето случаи, по неколку недели или месеци - целосно закрепнување, понекогаш - резидуални нарушувања во форма на лесна церебеларна инсуфициенција.
Мари-Фокс-Алахуанинова болест - доцна симетрична кортикална атрофија на малиот мозок со доминантна лезија на пириформните неврони (пуркиниевите клетки) и грануларниот слој на кортексот, како и оралниот дел на церебеларниот вермис и дегенерација на маслинки. Се манифестира кај лица на возраст од 40-75 години со нарушување на рамнотежата, атаксија, нарушување на одењето, нарушувања на координацијата и намален мускулен тонус, главно на нозете; намерниот тремор во рацете не е многу изразен. Можни се нарушувања на говорот, но не припаѓаат на задолжителните знаци на болеста. Болеста беше опишана во 1922 година од француските невропатолози P. Marie, Ch. Фоикс и Т. Алаџуанин. Болеста е спорадична. Етиологијата на болеста не е разјаснета. Постојат мислења за провоцирачката улога на интоксикација, првенствено злоупотреба на алкохол, како и хипоксија, наследен товар. Клиничката слика е потврдена со КТ податоци на главата, што открива изразено намалување на волуменот на малиот мозок наспроти позадината на дифузните атрофични процеси во мозокот. Покрај тоа, високото ниво на аминотрансферази во крвната плазма се препознава како карактеристично (Пономарева Е.Н. et al., 1997).
