7.1. STRUKTUR, TILKOBLINGER OG FUNKSJONER AV CEREBEL
Lillehjernen (hjernen) ligger under en duplikat dura mater kjent som omrisset av lillehjernen(tentorium cerebelli), som deler kraniehulen i to ulike rom - supratentorial og subtentorial. V subtentorial plass, bunnen av denne er den bakre kraniale fossa, i tillegg til lillehjernen, er hjernestammen. Volumet av lillehjernen er i gjennomsnitt 162 cm 3. Vekten varierer mellom 136-169 g.
Lillehjernen ligger over broen og medulla oblongata. Sammen med de overlegne og underordnede hjerneseilene utgjør den taket til den fjerde ventrikkelen i hjernen, hvis bunn er den såkalte romboide fossa (se kapittel 9). Over lillehjernen er oksipitallappene i den store hjernen, atskilt fra den av lillehjernens tentorium.
I lillehjernen er det to halvkuler(hemispherum cerebelli). Mellom dem, i sagittalplanet over IV-ventrikkelen i hjernen, ligger den fylogenetisk eldste delen av lillehjernen - dens mark(vermis cerebelli). Vermis og cerebellar hemisfærer er fragmentert i lobuler av dype tverrgående riller.
Lillehjernen er sammensatt av grå og hvit substans. Den grå substansen danner cerebellar cortex og de parede nuclei nuclei cerebelli lokalisert i dens dybde (fig. 7.1). Den største av dem er taggete kjerner(nucleus dentatus) - ligger i halvkulene. I den sentrale delen av ormen er det teltkjerner(kjerner
Ris. 7.1. Lillehjernekjerner.
1 - tannkjerne; 2 - korkaktig kjerne; 3 - kjernen av teltet; 4 - sfærisk kjerne.
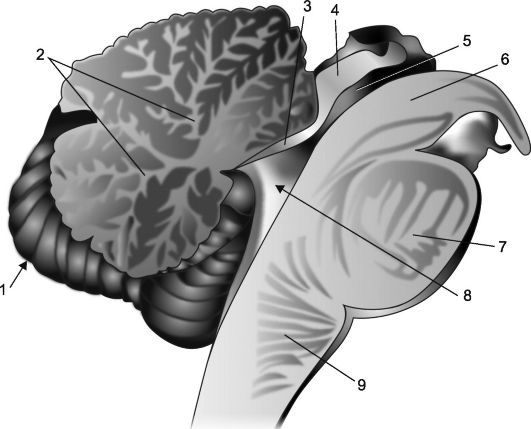
Ris. 7.2.Sagittal del av lillehjernen og hjernestammen.
1 - lillehjernen; 2 - "livets tre"; 3 - forhjerneseil; 4 - plate av firemannsrom; 5 - akvedukt av hjernen; 6 - hjernens ben; 7 - bro; 8 - IV ventrikkel, dens choroid plexus og telt; 9 - medulla oblongata.
fastigii), mellom dem og dentate kjernene er sfærisk og korkete kjerner(nuctei.globosus et emboliformis).
På grunn av det faktum at cortex dekker hele overflaten av lillehjernen og trenger inn i dypet av furene, på en sagittal del av lillehjernen, har vevet et bladmønster, hvis årer er dannet av en hvit substans (fig. 7.2), som utgjør den såkalte livets tre i lillehjernen (arbor vitae cerebelli). Ved bunnen av livets tre er det et kileformet hakk, som er den øvre delen av hulrommet til IV-ventrikkelen; kantene på denne fordypningen danner teltet hans. Taket på teltet er cerebellar ormen, og dens fremre og bakre vegger er tynne hjerneplater kjent som fremre og bakre. hjernen seiler(vella medullare anterior et posterior).
Litt informasjon om cerebellar arkitektonikk, gi grunnlag for å bedømme funksjonen til dens komponenter. Ha cerebellar cortex Det er to cellelag: det indre er granulært, bestående av små kornceller, og det ytre er molekylært. Mellom dem er det en rekke store pæreformede celler som bærer navnet til den tsjekkiske vitenskapsmannen I. Purkinje som beskrev dem (Purkinje I., 1787-1869).
Impulsene kommer inn i cerebellar cortex gjennom mosegrodde og krypende fibre som trenger inn i den fra den hvite substansen, som utgjør de afferente banene til cerebellum. Gjennom mosete fibre, impulser fra ryggmargen
vestibulære kjerner og kjerner av pons overføres til cellene i det granulære laget av cortex. Aksonene til disse cellene, sammen med krypende fibre som passerer gjennom det granulære laget i transitt og bærer impulser fra de underordnede olivenene til lillehjernen, når det overfladiske, molekylære laget av lillehjernen. Her deler aksonene til cellene i det granulære laget og de krypende fibrene seg i en T-form, og i det molekylære laget tar grenene deres en retning langsgående til overflaten av lillehjernen. Impulsene som har nådd det molekylære laget av cortex, etter å ha passert gjennom de synaptiske kontaktene, faller på de forgrenende dendrittene til Purkinje-celler som ligger her. Deretter følger de dendrittene til Purkinje-celler til kroppene deres, som ligger på grensen til de molekylære og granulære lagene. Deretter, langs aksonene til de samme cellene som krysser det granulære laget, trenger de inn i dybden av den hvite substansen. Aksonene til Purkinje-celler ender i kjernene i lillehjernen. Hovedsakelig i dentate nucleus. Efferente impulser som kommer fra lillehjernen langs aksonene til cellene som utgjør kjernen og som deltar i dannelsen av lillehjernen, forlater lillehjernen.
Lillehjernen har tre par ben: bunn, midt og topp. Underbenet forbinder det med medulla oblongata, midten - med broen, den øvre - med midthjernen. Hjernens ben utgjør banene som bærer impulser til og fra lillehjernen.
Cerebellar vermis sikrer stabilisering av kroppens tyngdepunkt, dens balanse, stabilitet, regulering av tonen til gjensidige muskelgrupper, hovedsakelig nakke og kropp, og fremveksten av fysiologiske cerebellare synergier som stabiliserer balansen i kroppen.
For å lykkes med å opprettholde balansen i kroppen mottar lillehjernen konstant informasjon som passerer langs spinocerebellarbanene fra proprioseptorene til forskjellige deler av kroppen, så vel som fra de vestibulære kjernene, underordnede oliven, den retikulære formasjonen og andre formasjoner som er involvert i å kontrollere plassering av kroppsdeler i rommet. De fleste av de afferente banene som fører til lillehjernen passerer gjennom den nedre lillehjernens pedikel, noen av dem er lokalisert i den overordnede lillehjernens pedikel.
Impulser av proprioseptiv følsomhet, å gå til lillehjernen, som andre sensoriske impulser, etter dendrittene til de første sensoriske nevronene, når kroppene deres som ligger i spinalknutene. Deretter blir impulser som går til lillehjernen langs aksonene til de samme nevronene rettet til kroppene til andre nevroner, som er lokalisert i de indre delene av bunnen av de bakre hornene, og danner den såkalte Clarks søyler. Aksonene deres faller inn i laterale seksjoner av laterale ledninger i ryggmargen, hvor de danner spinocerebellare veier, i dette tilfellet faller en del av aksonene inn i sidesøylen på samme side og dannes der den bakre spinocerebellar-kanalen til Flexig (tractus spinocerebellaris posterior). En annen del av aksonene til cellene i de bakre hornene passerer til den andre siden av ryggmargen og går inn i den motsatte sidemargen og dannes i den fremre spinocerebellar-kanal hos Govers (tractus spinocerebellaris anterior). Spinocerebellar-kanalene, øker i volum på nivået av hvert spinalsegment, stiger til medulla oblongata.
I medulla oblongata avviker den bakre spinocerebellarbanen i lateral retning, og etter å ha passert gjennom den nedre cerebellar pedicle, trenger den inn i cerebellum. Den fremre spinocerebellare banen passerer gjennom medulla oblongata, hjernens pons, og når midthjernen, på nivået som den gjør sitt andre skjæringspunkt i fremre cerebral velum og passerer inn i cerebellum gjennom den overordnede lillehjernens peduncle.
Således, av de to spinalkanalene, krysses den ene aldri (ikke krysset Fleksigs vei), og den andre passerer til motsatt side to ganger (to ganger krysset av Govers). Som et resultat leder begge impulser fra hver halvdel av kroppen, hovedsakelig til den homolaterale halvdelen av lillehjernen.
I tillegg til Fleksigs spinocerebellar tractus, passerer impulser til cerebellum gjennom den nedre cerebellar pedicle langs vestibulocerebellarkanalen (tractus vestibulocerebellaris), starter hovedsakelig i den øvre vestibulære kjernen av ankyloserende spondylitt, og langs olivo-cerebellarkanalen (tractus olivocerebellaris) som kommer fra den nedre oliven. En del av aksonene til cellene til de tynne og kileformede kjernene, ikke deltar i dannelsen av bulbothalamic-kanalen, i form av eksterne buede fibre (fiber arcuatae externae) går også inn i lillehjernen gjennom den nedre lillehjernens peduncle.
Gjennom mellombena mottar lillehjernen impulser fra hjernebarken. Disse impulsene går gjennom kortikale-cerebellopontine-baner, bestående av to nevroner. Kroppene til de første nevronene er lokalisert i hjernebarken, hovedsakelig i cortex av de bakre delene av frontallappene. Aksonene deres passerer som en del av den strålende kronen, den fremre delen av den indre kapselen og ender i brokjernene. Aksoner av celler av andre nevroner, hvis kropper er lokalisert i sine egne brokjerner, gå til motsatt side og lag opp, etter krysset, den midtre lillehjernens pedikel,
ender i motsatt halvkule av lillehjernen.
Noen av impulsene som har oppstått i hjernebarken når den motsatte halvkulen av lillehjernen, og gir informasjon ikke om den produserte, men bare om den planlagte aktive bevegelsen. Etter å ha mottatt slik informasjon, lillehjernen sender øyeblikkelig ut impulser som korrigerer frivillige bevegelser, hovedsakelig, ved å slukke treghet og det mest rasjonelle regulering av gjensidig muskeltonus - muskelagonister og -antagonister. Som et resultat, en slags eimetri, gjøre frivillige bevegelser klare, perfeksjonerte, blottet for upassende komponenter.
Banene som forlater lillehjernen er sammensatt av aksonene til cellene, hvis kropper danner kjernene. De fleste efferente veier, inkludert veier fra dentate kjernene, forlate lillehjernen gjennom øvre ben. På nivået av de nedre tuberklene til firedoblingen krysser den efferente cerebellarkanalen (skjæringspunktet mellom de øvre cerebellarbena til Werneking). Etter å ha krysset hver og en av dem når de røde kjernene på motsatt side av midthjernen. I de røde kjernene bytter lillehjernens impulser til neste nevron og beveger seg deretter langs aksonene til cellene, hvis kropper er innebygd i de røde kjernene. Disse aksonene er dannet i rød-ryggradsveier (tracti rubro spinalis), stiene til Monakov, som like etter utganger fra røde kjerner gjennomgår en kryssing (dekkkryss eller ørretkryss), hvoretter de går ned i ryggmargen. I ryggmargen er de rød-nukleære ryggmargene lokalisert i sidemargene; deres bestanddeler ender ved cellene i de fremre hornene i ryggmargen.
Hele den efferente veien fra lillehjernen til cellene i de fremre hornene i ryggmargen kan kalles lillehjernen-rød-kjernefysisk-ryggrad (tractus cerebello-rubrospinalis). Han krysser to ganger (skjæringspunktet mellom de overordnede lillehjernens peduncles og skjæringspunktet mellom operculum) og forbinder til slutt hver cerebellar hemisfære med perifere motoriske nevroner lokalisert i de fremre hornene til den homolaterale halvdelen av ryggmargen.
Fra kjernene til cerebellar vermis går de efferente banene hovedsakelig gjennom den nedre cerebellar pedikelen til den retikulære dannelsen av hjernestammen og de vestibulære kjernene. Herfra, langs de retikulospinale og vestibulospinale banene som går langs de fremre ledningene av ryggmargen, når de også cellene i de fremre hornene. En del av impulsene som kommer fra lillehjernen, som passerer gjennom de vestibulære kjernene, går inn i den mediale langsgående bunten, når kjernene III, IV og VI til kranialnervene som gir øyeeplenes bevegelse, og påvirker deres funksjon.
Oppsummert bør følgende vektlegges:
1. Hver halvdel av lillehjernen mottar hovedsakelig impulser a) fra den homolaterale halvdelen av kroppen, b) fra den motsatte hjernehalvdelen, som har kortiko-spinalforbindelser med samme halvdel av kroppen.
(2) Fra hver halvdel av lillehjernen ledes efferente impulser til cellene i de fremre hornene i den homolaterale halvdelen av ryggmargen og til kjernene i kranienervene som gir bevegelse av øyeeplene.
Denne arten av cerebellarforbindelsene gjør det mulig å forstå hvorfor, når den ene halvdelen av cerebellum er påvirket, forekommer cerebellare lidelser hovedsakelig i samme, dvs. homolateral, halvparten av kroppen. Dette er spesielt uttalt når de lille hjernehalvdelene er påvirket.
7.2. FORSKNING AV FUNKSJONENE TIL CEREBELLA
OG KLINISKE MANIFESTASJONER AV DERES NEDERLAG
Med skade på lillehjernen er forstyrrelser av statikk og koordinering av bevegelser, muskelhypotoni og nystagmus karakteristiske.
Cerebellar lesjon først av alt ormen hans, fører til brudd på statikk - evnen til å opprettholde en stabil posisjon av tyngdepunktet til menneskekroppen, balanse, stabilitet. Når denne funksjonen er forstyrret, statisk ataksi (fra gresk ataksi - uorden, ustabilitet). Ustabiliteten til pasienten er notert. Derfor, i stående stilling, sprer han bena langt fra hverandre, balanserer med hendene. Spesielt tydelig statisk ataksi oppdages med en kunstig reduksjon i støtteområdet, spesielt i Romberg-stillingen. Pasienten inviteres til å reise seg, bevege føttene bestemt og løfte hodet lett. I nærvær av cerebellare lidelser noteres pasientens ustabilitet i denne posisjonen, kroppen hans svaier, noen ganger "trekker" den i en bestemt retning, og hvis pasienten ikke støttes, kan han falle. Ved skade på cerebellar ormen svaier pasienten vanligvis fra side til side og faller ofte tilbake. Med patologi av cerebellar hemisfære, er det en tendens til å falle hovedsakelig mot det patologiske fokuset. Hvis den statiske lidelsen er moderat uttrykt, er det lettere å identifisere i den såkalte komplisert eller sensibilisert Romberg-positur. Pasienten blir bedt om å sette føttene i en linje slik at tåen på den ene foten hviler på hælen på den andre. Stabilitetsvurderingen er den samme som i vanlig Romberg-stilling.
Normalt, når en person står, er musklene i bena hans spente. (støttereaksjon), med trusselen om å falle til siden, beveger benet hans på denne siden i samme retning, og det andre benet kommer fra gulvet (hoppreaksjon). Ved skade på lillehjernen (hovedsakelig ormen) blir pasientens reaksjoner forstyrret
støtte og hoppe. Brudd på støttereaksjonen manifesteres av pasientens ustabilitet i stående stilling, spesielt i Romberg-stillingen. Brudd på hoppreaksjonen fører til det faktum at hvis legen, som står bak pasienten og forsikrer ham, skyver pasienten i en eller annen retning, så faller pasienten med et lett trykk (skyvesymptom).
Ved skade på lillehjernen endres vanligvis pasientens gang på grunn av utviklingen statomotorisk ataksi. Lillehjernens gang på mange måter ligner ganglaget til en full person, derfor kalles det noen ganger "en fulls ganglag." På grunn av ustabilitet går pasienten usikker, sprer bena langt fra hverandre, mens han «kastes» fra side til side. Og når lillehjernen er skadet, avviker den når man går fra en gitt retning mot det patologiske fokuset. Ustabiliteten er spesielt uttalt ved svinger. Hvis ataksien er uttalt, mister pasientene fullstendig evnen til å kontrollere kroppen sin og kan ikke bare stå og gå, men til og med sitte.
Den dominerende lesjonen i cerebellare hemisfærer fører til en forstyrrelse av dens anti-tregasjonseffekter, spesielt til fremveksten kinetisk ataksi. Det manifesteres av kjeftheten i bevegelser og er spesielt uttalt med bevegelser som krever presisjon. For å oppdage kinetisk ataksi utføres tester for koordinering av bevegelser. Noen av dem er beskrevet nedenfor.
Test for diadokokinese (fra gresk. diadochos - sekvens). Pasienten inviteres til å lukke øynene, strekke armene fremover og raskt, rytmisk supinere og penetrere hendene. Ved skade på cerebellar hemisfære, viser håndbevegelsene på siden av den patologiske prosessen seg å være mer feiende (en konsekvens av dysmetri, mer presist, hypermetri), som et resultat begynner hånden å henge etter. Dette indikerer tilstedeværelsen av adiadokokinesis.
Fingertest. En pasient med lukkede øyne skal trekke hånden, og deretter sakte, med pekefingeren, berøre nesetippen. I tilfelle av cerebellar patologi, gjør hånden på siden av det patologiske fokuset en overdreven bevegelse i volum (hypermetri), som et resultat av at pasienten savner. En finger-nese-test avslører en karakteristikk av cerebellar patologi cerebellar (tilsiktet) tremor, hvis amplitude øker når fingeren nærmer seg målet. Denne testen lar deg identifisere den såkalte bradytelekinesien. (hodelagssymptom): ikke langt fra målet bremses fingerens bevegelse, noen ganger til og med pauser, for så å fortsette igjen.
Finger-finger test. En pasient med lukkede øyne inviteres til å spre armene bredt og deretter bringe pekefingrene nærmere hverandre, og prøve å få fingeren inn i fingeren, mens det, som med fingertesten, avsløres en tilsiktet skjelving og et hodelagssymptom.
Calcaneal kne test (fig. 7.3). Pasienten, liggende på ryggen med lukkede øyne, blir bedt om å heve det ene benet høyt og deretter slå kneet på det andre benet med hælen. Med cerebellar patologi kan pasienten ikke eller det er vanskelig for ham å få hælen inn i kneet på det andre benet, spesielt når du utfører en test med benet homolateralt til den berørte cerebellar hemisfæren. Hvis hælen likevel når kneet, foreslås det å holde den, lett berører den fremre overflaten av underbenet, ned til ankelleddet, mens i tilfelle av cerebellar patologi glir hælen av underbenet hele tiden i den ene eller andre retningen.

Ris. 7.3.Calcaneal kne test.
Veiledende test: Pasienten inviteres til å slå gummituppen på hammeren, som er i undersøkerens hånd, med pekefingeren flere ganger. Ved cerebellar patologi i pasientens hånd på siden av den berørte cerebellar hemisfæren er det en feilstilling på grunn av dysmetri.
Tom-Jumenti symptom: Hvis pasienten tar opp en gjenstand, for eksempel et glass, sprer han fingrene for mye.
Lillehjernens nystagmus. Rykninger i øyeeplene når man ser til sidene (horisontal nystagmus) anses som en konsekvens av tilsiktet skjelving av øyeeplene (se kapittel 30).
Taleforstyrrelser: Tale mister sin flyt, blir eksplosiv, fragmentert, sang som cerebellar dysartri (se kapittel 25).
Endre håndskrift: På grunn av forstyrrelsen i koordineringen av håndbevegelser, blir håndskriften ujevn, bokstavene er deformerte, for store (megalografi).
Pronatorisk fenomen: Pasienten blir bedt om å holde armene utstrakt i supinasjonsstilling, mens spontan pronasjon snart oppstår på siden av den berørte lillehjernen.
Hoff-Schilder symptom: Hvis pasienten holder armene strakt fremover, og på siden av den berørte halvkulen, trekkes armen snart utover.
Et imitasjonsfenomen. En pasient med lukkede øyne bør raskt gi hånden en stilling som ligner den som undersøkeren tidligere hadde gitt den andre hånden. Når lillehjernen er skadet, gjør den homolaterale hånden en bevegelse som er for stor i amplitude.
Doinikovs fenomen. Fingerfenomen. Den sittende pasienten inviteres til å legge supinerte hender med fingrene fra hverandre på lårene og lukke øynene. Ved en lesjon av lillehjernen på siden av det patologiske fokuset oppstår det snart spontanfleksjon av fingrene og pronasjon av hånd og underarm.
Stuart-Holmes symptom. Undersøkeren ber pasienten som sitter på stolen om å bøye de supinerte underarmene og samtidig, ta hendene i håndleddene, motstå ham. Hvis du samtidig plutselig slipper pasientens hender, vil hånden på den berørte siden, bøyd av treghet, treffe ham kraftig i brystet.
Muskel hypotensjon. Nederlaget til cerebellar vermis fører vanligvis til diffus muskelhypotensjon. Med nederlaget til cerebellar hemisfære avslører passive bevegelser en reduksjon i muskeltonus på siden av den patologiske prosessen. Muskelhypotoni fører til muligheten for hyperekstensjon av underarmen og underbenet (Olshansky symptom) med passive bevegelser, til utseendet symptomer på en hengende hånd eller fot med deres passive risting.
Patologiske cerebellare asynergier. Brudd på fysiologiske synergier under komplekse motoriske handlinger avsløres, spesielt under følgende tester (fig. 7.4).
1. Asynergi ifølge Babinsky i stående stilling. Hvis en pasient som står med forskjøvede ben prøver å bøye seg bakover og kaste hodet bakover, oppstår normalt i dette tilfellet fleksjon av kneleddene. I cerebellar patologi på grunn av asynergi, er denne vennlige bevegelsen fraværende, og pasienten, mister balansen, faller tilbake.
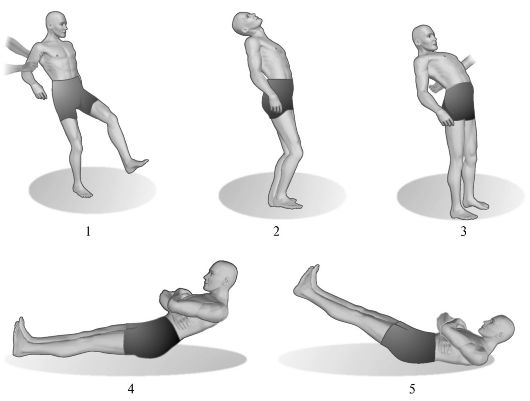
Ris. 7.4.Cerebellar asynergi.
1 - gangart til en pasient med alvorlig cerebellar ataksi; 2 - tilbakehelling av kroppen er normal; 3 - med skade på lillehjernen, kan pasienten, som bøyer seg bakover, ikke opprettholde balansen; 4 - utføre en test for cerebellar asynergi i henhold til Babinsky av en sunn person; 5 - utfører den samme testen hos pasienter med cerebellare lesjoner.
2. Asynergi ifølge Babinsky i ryggleie. Pasienten, liggende på et fast plan med bena utstrakt, spredt til bredden av skulderbeltet, inviteres til å krysse armene over brystet og deretter sette seg ned. I nærvær av cerebellar patologi på grunn av fraværet av vennlig sammentrekning av glutealmusklene (manifestation av asynergi), kan pasienten ikke fikse bena og bekkenet på støtteområdet, som et resultat, bena stiger og han kan ikke sette seg ned. Betydningen av dette symptomet bør ikke overvurderes hos eldre pasienter, hos personer med slapp eller overvektig bukvegg.
For å oppsummere det ovenstående, bør mangfoldet og viktigheten av funksjonene som utføres av lillehjernen understrekes. Som en del av en komplekssme, fungerer lillehjernen som et fokuspunkt for å balansere kroppen og opprettholde muskeltonen. Som P. Duus (1995) bemerker, lillehjernen gir evnen til å utføre diskrete og presise bevegelser, Forfatteren tror med rimelighet at lillehjernen fungerer som en datamaskin, sporer og koordinerer sensorisk informasjon ved inngangen og simulerer motoriske signaler ved utgangen.
7.3. MULTISYSTEM DEGENERASJONER
Med tegn på cerebellar patologi
Multisystemdegenerasjoner er en gruppe nevrodegenerative sykdommer, hvor fellestrekket er lesjonens multifokale natur med involvering av forskjellige funksjonelle og nevrotransmittersystemer i hjernen i den patologiske prosessen og derfor den polysystemiske karakteren til kliniske manifestasjoner.
7.3.1. Cerebellar ataksi
Spinocerebellar ataksier inkluderer progressive arvelige degenerative sykdommer, der strukturene i lillehjernen, hjernestammen og ryggmargsbanene, som hovedsakelig er relatert til det ekstrapyramidale systemet, hovedsakelig påvirkes.
7.3.1.1. Friedreichs arvelige ataksi
Arvelig sykdom beskrevet i 1861 av den tyske nevropatologen N. Friedreich (Friedreich N., 1825-1882). Det arves på en autosomal recessiv måte eller (sjeldnere) på en autosomal dominant måte med ufullstendig penetrans og variabel genuttrykk. Sporadiske tilfeller av sykdommen er også mulig.
Patogenesesykdom ikke spesifisert. Spesielt er det ingen anelse om den primære biokjemiske defekten som utgjør grunnlaget.
Patomorfologi.Patologiske studier viser en uttalt tynning av ryggmargen på grunn av atrofiske prosesser i dens bakre og laterale ledninger. Som regel lider de kileformede (Burdach) og milde (Gaulle) banene og spinocerebellarbanene til Govers og Fleksig, samt den kryssede pyramidebanen som inneholder
mange fibre relatert til det ekstrapyramidale systemet. Degenerative prosesser kommer også til uttrykk i lillehjernen, i dens hvite substans og i kjernefysisk apparat.
Kliniske manifestasjoner. Sykdommen viser seg hos barn eller unge under 25 år. S.N. Davidenkov (1880-1961) bemerket at de kliniske tegnene på sykdommen oftere forekommer hos barn i alderen 6-10 år. Det første tegn på sykdom er vanligvis ataksi. Pasienter opplever usikkerhet, vaklende når de går, endringer i gange (når de går, er bena vidt fra hverandre). Gangen ved Friedreichs sykdom kan kalles tabetisk-cerebellar, siden forandringene er forårsaket av en kombinasjon av sensitiv og cerebellar ataksi, samt en vanligvis uttalt reduksjon i muskeltonus. Forstyrrelser av statikk, diskordinasjon i hendene, tilsiktet tremor, dysartri er også karakteristiske. Mulig nystagmus, hørselstap, elementer av chanting tale, tegn på pyramidal insuffisiens (senehyperrefleksi, fotpatologiske reflekser, noen ganger en liten økning i muskeltonus), imperativ trang til å urinere, nedsatt seksuell potens. Noen ganger oppstår athetoid hyperkinesi.
En tidlig debuterende lidelse med dyp følsomhet fører til en progressiv reduksjon i senereflekser: først på bena og deretter på armene. Over tid dannes muskelhypotrofi i de distale bena. Tilstedeværelsen av anomalier i utviklingen av skjelettet er karakteristisk. Først av alt manifesteres dette av tilstedeværelsen Friedreichs føtter: foten er forkortet, "hul", med en veldig høy bue. Hovedfalangene til fingrene hennes er ubøyde, resten er bøyd (fig. 7.5). Mulig deformasjon av ryggraden, brystet. Det er ofte manifestasjoner av kardiopati. Sykdommen utvikler seg sakte, men fører stadig til uførhet hos pasienter som til slutt blir sengeliggende.
Behandling. Patogenetisk behandling er ikke utviklet. Foreskrive legemidler som forbedrer metabolismen i nervesystemets strukturer, styrkende midler. Med alvorlig deformitet av føttene er ortopediske sko indikert.
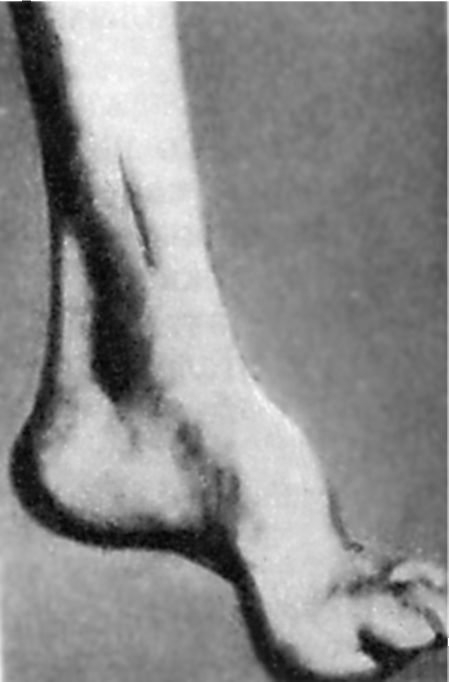
Ris. 7.5.Friedreichs fot.
7.3.1.2. Arvelig cerebellar ataksi (Pierre Maries sykdom)
Dette er en kronisk progressiv arvelig sykdom, manifestert i en alder av 30-45 år, med sakte voksende cerebellare lidelser i kombinasjon med tegn på pyramidal insuffisiens, mens statisk og dynamisk cerebellar ataksi, tilsiktet skjelving, chanted tale, senehyperrefleksi er karakteristiske. Mulige kloner, patologiske pyramidale reflekser, strabismus, nedsatt syn, innsnevring av synsfeltene på grunn av primær atrofi av synsnervene og pigmentær degenerasjon av netthinnen. Sykdomsforløpet er sakte progressivt. Det er en reduksjon i størrelsen på lillehjernen, celledegenerasjon
Purkinje, mindreverdige oliven, spinocerebellar kanaler. Det arves på en autosomal dominant måte. Sykdommen ble beskrevet i 1893 av den franske nevropatologen R. Marie (1853-1940).
Foreløpig er det ingen enstemmighet i forståelsen av begrepet «Pierre Maries sykdom», og spørsmålet om muligheten for å skille det ut i en uavhengig nosologisk form kan diskuteres.
Ingen behandling er utviklet. Vanligvis brukes metabolsk aktive og gjenopprettende, samt symptomatiske midler.
7.3.2. Olivopontocerebellar dystrofi (Dejerine-Thom sykdom)
Dette er en gruppe kroniske, progressive arvelige sykdommer, der dystrofiske forandringer utvikler seg hovedsakelig i lillehjernen, nedre oliven, i ponsens egne kjerner og i hjernestrukturene knyttet til dem.
Ved utvikling av sykdommen i ung alder arves omtrent halvparten av tilfellene på en dominant eller recessiv måte, resten er sporadiske. I sporadiske tilfeller av sykdommen er manifestasjoner av akinetisk-rigid syndrom og progressiv autonom svikt mer vanlig. Gjennomsnittsalderen til pasienten med manifestasjonen av den arvelige formen av sykdommen i fenotypen er 28 år, med sporadiske - 49 år, er gjennomsnittlig levealder henholdsvis 14,9 og 6,3 år. I den sporadiske formen, i tillegg til atrofi av oliven, pons og lillehjernen, er det oftere funnet lesjoner i laterale ledninger i ryggmargen, substantia nigra og striatum, en blåaktig flekk i rhomboid fossa i den fjerde ventrikkelen i hjernen. .
Symptomer på voksende cerebellar syndrom er karakteristiske. Følsomhetsforstyrrelser, elementer av bulbare og akinetisk-stive syndromer, hyperkinese, spesielt myorrytmier i tungen og den myke gane, oftalmoparese, nedsatt synsskarphet, intellektuelle forstyrrelser er mulig. Sykdommen ble beskrevet i 1900 av de franske nevropatologene J. Dejerine og A. Thomas.
Sykdommen debuterer ofte med forstyrrelser i gange - ustabilitet, diskordinasjon, uventede fall er mulig. Disse lidelsene kan være den eneste manifestasjonen av sykdommen innen 1-2 år. I fremtiden oppstår og vokser koordinasjonsforstyrrelser i hendene: manipulasjoner med små gjenstander er vanskelige, håndskrift forstyrres, en forsettlig skjelving oppstår. Talen blir intermitterende, uskarp, med et neseskjær og pusterytmen som ikke samsvarer med talens struktur (pasienten snakker som om han blir kvalt). På dette stadiet av sykdommen slutter manifestasjoner av progressiv autonom insuffisiens seg, tegn på akinetisk-rigid syndrom vises. Noen ganger er de dominerende symptomene for pasienten dysfagi, angrep av nattlig kvelning. De utvikles i forbindelse med blandede parese av bulbarmusklene og kan være livstruende.
I 1970 ble de tyske nevropatologene B.W. Konigsmark og L.P. Weiner trakk seg ut 5 hovedtyper olivopontocerebellar dystrofi, forskjellig enten i kliniske og morfologiske manifestasjoner, eller i type arv.
Jeg type (Menzel type). I en alder av 14-70 (oftere 30-40) år manifesterer det seg ataksi, dysartri, dysfoni, muskelhypotoni, i det sene stadiet - en kraftig skjelving i hodet, bagasjerommet, armene, muskler, tegn på akinetisk- rigid syndrom. Mulige patologiske pyramidale tegn, blikkpareser, ekstern og intern oftalmoplegi, sensitivitetsforstyrrelser, demens. Det arves på en autosomal dominant måte. Den ble skilt ut som en uavhengig form i 1891 av P. Menzel.
II type (Fickler-Winkler type) ... I en alder av 20-80 viser det seg ataksi, nedsatt muskeltonus og senereflekser. Det arves på en autosomal recessiv måte. Sporadiske tilfeller er mulige.
III type med retinal degenerasjon. Det manifesterer seg i barndom eller ung (opptil 35 år) alder ataksi, skjelving i hodet og ekstremiteter, dysartri, tegn på pyramidal insuffisiens, progressiv reduksjon i synet med utfall i blindhet; mulig nystagmus, oftalmoplegi, noen ganger dissosierte sensoriske forstyrrelser. Det arves på en autosomal dominant måte.
IV type (Jester-Highmaker type). I en alder av 17-30 år debuterer han med cerebellar ataksi eller tegn på nedre spastisk paraparese, i begge tilfeller, allerede på et tidlig stadium av sykdommen, dannes en kombinasjon av disse manifestasjonene, til hvilke elementer av bulbar syndrom, parese av ansiktsmuskulaturen, og dype sensitivitetsforstyrrelser blir senere lagt til. Dominant arvet.
V type. Det manifesterer seg i en alder av 7-45 år ataksi, dysartri, tegn på akinetic-rigid syndrom og andre ekstrapyramidale lidelser, progressiv oftalmoplegi og demens er mulig. Dominant arvet.
7.3.3. Olivorubrocerebellar degenerasjon (Lejeune-Lermitte syndrom, Lermitte sykdom)
Sykdommen er preget av progressiv atrofi av lillehjernen, hovedsakelig av dens cortex, dentate kjerner og øvre cerebellar peduncles, inferior oliven og røde kjerner. Det manifesteres først og fremst av statisk og dynamisk ataksi; i fremtiden er andre tegn på cerebellar syndrom og skade på hjernestammen mulig. Sykdommen ble beskrevet av de franske nevropatologene J. Lhermitte (Lhermitte J.J., 1877-1959) og J. Lezhon (Lejonne J., født i 1894).
7.3.4. Multisystematrofi
I de siste tiårene har en sporadisk, progressiv nevrodegenerativ sykdom kalt multisystematrofi blitt identifisert som en uavhengig form. Det er preget av en kombinert lesjon av basalgangliene, lillehjernen, hjernestammen, ryggmargen. De viktigste kliniske manifestasjonene: parkinsonisme, cerebellar ataksi, tegn på pyramidal og autonom insuffisiens (Levin O.S., 2002). Avhengig av overvekten av visse trekk ved det kliniske bildet, skilles tre typer multisystematrofi.
1) olivopontocerebellar type, preget av overvekt av tegn på cerebellar angrep;
2) strionigral type, der tegn på parkinsonisme dominerer;
3) Shai-Drager syndrom, karakterisert ved overvekt i det kliniske bildet av tegn på progressiv autonom svikt med symptomer på ortostatisk arteriell hypotensjon.
Grunnlaget for multisystematrofi er selektiv degenerasjon av visse områder av den overveiende grå substansen i hjernen med skade på nevroner og gliaelementer. Årsakene til degenerative manifestasjoner i hjernevevet er fortsatt ukjente i dag. Manifestasjonene av multisystematrofi av typen olivopontocerebellar er assosiert med skade på Purkinje-celler i cerebellar cortex, så vel som nevroner av underordnede oliven, pontocerebellare kjerner, demyelinisering og degenerasjon, hovedsakelig av pontocerebellarbanene.
Cerebellare lidelser er vanligvis statisk og dynamisk ataksi med nedsatt bevegelsesevne. Karakterisert av ustabilitet i Romberg-posisjonen, ataksi når du går, dysmetri, adiadokokinese, forsettlig skjelving, det kan være nystagmus (horisontal vertikal, nedslående), intermitterende og sakte sporende blikkbevegelser, nedsatt konvergens av øynene, chanted tale.
Multisystematrofi oppstår vanligvis i voksen alder og utvikler seg raskt. Diagnosen er basert på klinisk bevis og er preget av en kombinasjon av tegn på parkinsonisme, cerebellar svikt og autonome lidelser. Behandling av sykdommen er ikke utviklet. Varigheten av sykdommen - innen 10 år, ender med døden.
7.4. ANDRE SYKDOMMER ASSOSSIERT MED SYMPTOMER PÅ CEREBRAL SYKDOM
Hvis pasienten viser tegn på cerebellar lesjon, så i de fleste tilfeller, først og fremst du må tenke på muligheten cerebellare svulster(astrocytom, angioblastom, medulloblastom, metastatiske svulster) eller multippel sklerose. På cerebellare svulster tidlige tegn på intrakraniell hypertensjon vises. Ved multippel sklerose er det vanligvis mulig å identifisere, i tillegg til cerebellar patologi, kliniske manifestasjoner av skade på andre strukturer i sentralnervesystemet, først og fremst de visuelle og pyramidale systemene. I klassisk nevrologi er kjennetegn ved multippel sklerose Charcots triade: nystagmus, forsettlig skjelving og chanted tale, og Nonnes syndrom: forstyrrelse av koordinering av bevegelser, dysmetri, chanted tale og cerebellar asynergi.
Cerebellare lidelser er store og inn posttraumatisk Mann syndrom, som er preget av ataksi, diskordinasjon, asynergi, nystagmus. Traumer eller infeksjon kan forårsake cerebellar Goldstein-Reichmanns syndrom: forstyrrelser av statikk og koordinering av bevegelser, asynergi, forsettlig skjelving, nedsatt muskeltonus, hypermetri, megalografi, nedsatt oppfatning av massen (vekten) til et objekt i hendene.
Forstyrrelser i cerebellar funksjon kan også være medfødt i naturen, og manifestere seg spesielt, Zeemans syndrom: ataksi, forsinket taleutvikling, og deretter cerebellar dysartri.
Medfødt cerebellar ataksi Det manifesteres av en forsinkelse i utviklingen av barnets motoriske funksjoner (i en alder av 6 måneder kan han ikke sitte, begynner å gå sent, mens gangarten er ataktisk), samt forsinket tale, langvarig bevaring av dysartri, noen ganger mental retardasjon og mikrokranielle manifestasjoner er ikke uvanlig. På CT reduseres cerebellare hemisfærer. Ved ca 10 års alder skjer vanligvis kompensasjon av hjernefunksjoner, som imidlertid kan forstyrres under påvirkning av skadelige eksogene påvirkninger. Progressive former for sykdommen er også mulig.
En manifestasjon av medfødt hypoplasi av lillehjernen er og Fancony-Turner syndrom. Det er preget av nedsatt statikk og koordinering av bevegelser, nystagmus, som vanligvis er ledsaget av mental retardasjon.
Medfødt inkluderer også en autosomal recessiv arvelig type, som sjelden finnes Bettens sykdom: Det er preget av medfødt cerebellar ataksi, manifestert i det første leveåret av svekket statikk og koordinering av bevegelser, nystagmus, blikkkoordinasjonsforstyrrelse og moderat muskelhypotoni. Dysplastiske tegn er mulige. Barnet er sent, noen ganger bare ved 2-3 års alder, begynner å holde hodet, enda senere - å stå, gå, snakke. Talen hans endres i henhold til typen cerebellar dysartri. Vegetative-viscerale lidelser, manifestasjoner av immunsuppresjon er mulig. Etter noen år stabiliserer det kliniske bildet seg vanligvis, pasienten tilpasser seg til en viss grad de eksisterende defektene.
Spastisk ataksi etter forslag fra A. Bell og E. Carmichel (1939), ble cerebellar ataksi arvet av en autosomal dominant type navngitt, som er preget av sykdomsutbruddet ved 3-4 års alder og manifesteres av en kombinasjon av cerebellar. ataksi med dysartri, senehyperrefleksi og økt muskeltonus etter spastisk type, mens mulige (men ikke obligatoriske tegn på sykdommen) atrofi av synsnervene, retinal degenerasjon, nystagmus, oculomotoriske forstyrrelser.
Autosomal dominant er arvelig Feldman syndrom(beskrevet av den tyske legen H. Feldmann, født i 1919): cerebellar ataksi, tilsiktet skjelving og tidlig grånende hår. Det manifesterer seg i det andre tiåret av livet og går sakte videre, noe som fører til funksjonshemming etter 20-30 år.
Sen cerebellar atrofi eller Toms syndrom beskrevet i 1906 av den franske nevrologen A. Thomas (1867-1963), manifesterer seg vanligvis hos personer over 50 år med progressiv atrofi av cerebellar cortex. I fenotypen vises tegn på cerebellar syndrom, først og fremst cerebellar statisk og lokomotorisk ataksi, chanted tale og endringer i håndskrift. I et langt avansert stadium er manifestasjoner av pyramidal insuffisiens mulig.
Kombinasjonen av cerebellare lidelser med myoklonus er preget av Jakt myoklonisk cerebellar dyssynergi, eller myoklonus ataksi, med dette symptomkomplekset i det kliniske bildet, manifesteres tilsiktet skjelving, myoklonus som oppstår i hendene, og senere får en generalisert karakter, ataksi og dyssynergi, nystagmus, chanted tale, nedsatt muskeltonus. Det er en konsekvens av degenerasjon av cerebellarkjernene, røde kjerner og deres forbindelser, samt kortikale-subkortikale strukturer.
I et avansert stadium av sykdommen er epileptiske anfall og demens mulig. Prognosen er dårlig. Refererer til en sjelden form for progressiv arvelig ataksi. Det arves på en autosomal recessiv måte. Det vises vanligvis i ung alder. Den nosologiske uavhengigheten til symptomkomplekset er omstridt. Den amerikanske nevrologen R. Hunt (1872-1937) beskrev sykdommen i 1921.
Blant degenerative prosesser er et bestemt sted okkupert av Holmes cerebellar degenerasjon, eller familiær cerebellar atrofi, eller progressiv atrofi av cerebellarsystemet, hovedsakelig av dentate kjerner, samt røde kjerner, mens manifestasjoner av demyelinisering kommer til uttrykk i den superior cerebellar pedicle. Karakterisert av statisk og dynamisk ataksi, asynergi, nystagmus, dysartri, nedsatt muskeltonus, muskeldystoni, hodeskjelving, myoklonus. Epileptiske anfall vises nesten samtidig. Intelligens er vanligvis bevart. EEG viser paroksysmal dysrytmi. Sykdommen er anerkjent som arvelig, men arten av arv er ikke spesifisert. Beskrev sykdommen i 1907 av den engelske nevropatologen G. Holmes
(1876-1965).
Alkoholisk cerebellar degenerasjon - en konsekvens av kronisk alkoholforgiftning. Hovedsakelig er cerebellar-ormen påvirket, med cerebellar ataksi og nedsatt koordinering av benbevegelser som primært manifesteres, mens håndbevegelser, oculomotoriske og talefunksjoner er svekket i mye mindre grad. Vanligvis er denne sykdommen ledsaget av et uttalt hukommelsestap i kombinasjon med polynevropati.
manifesterer seg som cerebellar ataksi, som noen ganger kan være det eneste kliniske symptomet forbundet med en ondartet svulst, uten lokale tegn som indikerer stedet for dens forekomst. Paraneoplastisk cerebellar degenerasjon kan spesielt være en sekundær manifestasjon av bryst- eller eggstokkreft.
Barraquer-Bordas-Ruiz-Lara syndrom manifesterer seg som cerebellare lidelser som oppstår i forbindelse med raskt progressiv atrofi av lillehjernen. Et syndrom hos pasienter med bronkialkreft ledsaget av generell forgiftning er beskrevet av den moderne spanske legen L. Barraquer-Bordas (født i 1923).
Sjelden funnet recessiv X-kromosomal ataksi- en arvelig sykdom som manifesterer seg nesten bare hos menn med sakte progressiv cerebellar insuffisiens. Det overføres i en recessiv, kjønnsbundet type.
Bemerkelsesverdig og familiær paroksysmal ataksi, eller periodisk ataksi. Den debuterer oftere i barndommen, men den kan også dukke opp senere – opptil 60 år. Det kliniske bildet er redusert til paroksysmale manifestasjoner av nystagmus, dysartri og ataksi, nedsatt muskeltonus, svimmelhet, kvalme, oppkast, hodepine, som varer fra flere minutter til 4 uker.
Anfall av familiær paroksysmal ataksi kan utløses av emosjonelt stress, fysisk tretthet, feber, alkoholinntak, mens mellom angrepene blir fokale nevrologiske symptomer i de fleste tilfeller ikke oppdaget, men noen ganger er nystagmus og milde cerebellare symptomer mulig.
Det morfologiske substratet til sykdommen er anerkjent som en atrofisk prosess hovedsakelig i den fremre delen av cerebellarormen. Sykdommen ble først beskrevet i 1946 av M. Parker. Det arves på en autosomal dominant måte. I 1987, med familiær paroksysmal ataksi, ble det funnet en reduksjon i aktiviteten til pyruvatdehydrogenase av blodleukocytter til 50-60% av det normale nivået. I 1977 R. Lafrance et al. trakk oppmerksomhet til den høye profylaktiske effekten av diacarb, senere ble flunarizin foreslått for behandling av familiær paroksysmal ataksi.
Akutt cerebellar ataksi eller Leiden-Westphal syndrom, er et veldefinert symptomkompleks, som er en parainfeksiøs komplikasjon. Det forekommer oftere hos barn 1-2 uker etter en generell infeksjon (influensa, tyfus, salmonellose, etc.). Karakterisert av grov statisk og dynamisk ataksi, tilsiktet tremor, hypermetri, asynergi, nystagmus, chanted tale, nedsatt muskeltonus. I cerebrospinalvæsken påvises lymfocytisk pleocytose, en moderat økning i protein. Ved utbruddet av sykdommen er svimmelhet, bevissthetsforstyrrelser, kramper mulig. På CT og MR oppdages ikke patologi. Kurset er godartet. I de fleste tilfeller, etter noen uker eller måneder - fullstendig gjenoppretting, noen ganger - gjenværende lidelser i form av mild cerebellar insuffisiens.
Marie-Foix-Alajuanins sykdom - sen symmetrisk kortikal atrofi av lillehjernen med en dominerende lesjon av piriforme nevroner (Purkinje-celler) og det granulære laget av cortex, samt den orale delen av cerebellar vermis og olivendegenerasjon. Det viser seg hos personer i alderen 40-75 år med balanseforstyrrelser, ataksi, gangforstyrrelser, koordinasjonsforstyrrelser og nedsatt muskeltonus, hovedsakelig i bena; den tilsiktede skjelvingen i hendene er ikke særlig uttalt. Taleforstyrrelser er mulige, men hører ikke til de obligatoriske tegnene på sykdommen. Sykdommen ble beskrevet i 1922 av de franske nevropatologene P. Marie, Ch. Foix og Th. Alajouanine. Sykdommen er sporadisk. Etiologien til sykdommen er ikke avklart. Det er meninger om rusens provoserende rolle, først og fremst alkoholmisbruk, så vel som hypoksi, arvelig belastning. Det kliniske bildet bekreftes av CT-data fra hodet, som avslører en uttalt reduksjon i volumet av lillehjernen mot bakgrunnen av diffuse atrofiske prosesser i hjernen. I tillegg er et høyt nivå av aminotransferaser i blodplasma anerkjent som karakteristisk (Ponomareva E.N. et al., 1997).
