7.1. STRUCTURA, CONEXIUNI ȘI FUNCȚII ALE CEREBELULUI
Cerebelul (cerebelul) este situat sub o dura mater duplicată cunoscută ca conturul cerebelului(tentorium cerebelli), care împarte cavitatea craniană în două spații inegale - supratentorial și subtentorial. V spațiu subtentorial, al cărei fund este fosa craniană posterioară, pe lângă cerebel, este trunchiul cerebral. Volumul cerebelului este în medie de 162 cm 3. Greutatea sa variaza intre 136-169 g.
Cerebelul este situat deasupra podului și a medulului oblongata. Împreună cu pânzele cerebrale superioare și inferioare, constituie acoperișul celui de-al patrulea ventricul al creierului, al cărui fund este așa-numita fosă romboidă (vezi capitolul 9). Deasupra cerebelului se află lobii occipitali ai creierului mare, despărțiți de acesta de tentoriul cerebelului.
În cerebel, sunt două emisfere(emisferul cerebelului). Între ele, în planul sagital de deasupra ventriculului IV al creierului, se află cea mai veche parte filogenetic a cerebelului - sa vierme(vermis cerebel). Vermisul și emisferele cerebeloase sunt fragmentate în lobuli de șanțuri transversale profunde.
Cerebelul este compus din substanță cenușie și albă. Substanța cenușie formează cortexul cerebelos și nucleele pereche nucleele cerebelului situate în profunzimea sa (Fig. 7.1). Cele mai mari dintre ele sunt sâmburi zimțați(nucleus dentatus) – situat în emisfere. În partea centrală a viermelui există miezuri de cort(nuclee
Orez. 7.1. Nuclei cerebelosi.
1 - miez dintat; 2 - miez de plută; 3 - miezul cortului; 4 - nucleu sferic.
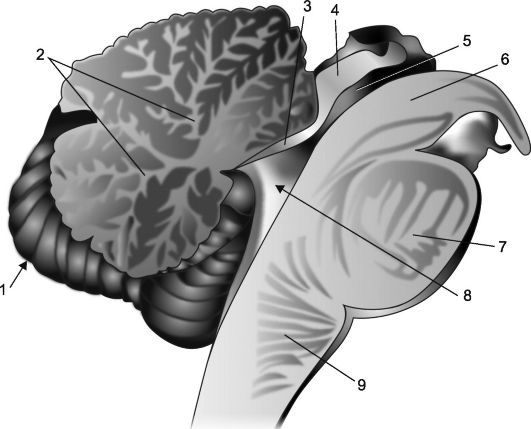
Orez. 7.2.Secțiunea sagitală a cerebelului și a trunchiului cerebral.
1 - cerebel; 2 - „arborele vieții”; 3 - velă prosenieră; 4 - farfurie de cvadruplu; 5 - apeductul creierului; 6 - piciorul creierului; 7 - pod; 8 - ventriculul IV, plexul coroid și cortul acestuia; 9 - medulla oblongata.
fastigii), între ele și nucleii dințați sunt sfericși nuclee de plută(nuctei.globosus et emboliformis).
Datorită faptului că cortexul acoperă întreaga suprafață a cerebelului și pătrunde în adâncurile brazdelor sale, pe o secțiune sagitală a cerebelului, țesutul său are un model de frunze, ale cărui vene sunt formate dintr-o substanță albă (Fig. . 7.2), care alcătuiește așa-numitul arborele vieții cerebelului (arbor vitae cerebelli). La baza arborelui vieții există o crestătură în formă de pană, care este partea superioară a cavității ventriculului IV; marginile acestei adâncituri formează cortul lui. Acoperișul cortului este viermele cerebelos, iar pereții săi anterior și posterior sunt plăci cerebrale subțiri cunoscute sub numele de anterioară și posterioară. pânzele creierului(vella medulară anterior și posterior).
Câteva informații despre arhitectura cerebeloasa, dând temeiuri pentru a judeca funcția componentelor sale. Avea cortexul cerebelos Există două straturi celulare: cel interior este granular, format din celule cu granule mici, iar cel exterior este molecular. Între ele se află o serie de celule mari în formă de pară care poartă numele savantului ceh I. Purkinje care le-a descris (Purkinje I., 1787-1869).
Impulsurile intră în cortexul cerebelos prin fibre mușchioase și târâtoare care pătrund în el din substanța albă, care alcătuiesc căile aferente ale cerebelului. Prin fibre mușchi, impulsuri din măduva spinării
nucleii vestibulari și nucleii pontului sunt transferați către celulele stratului granular al cortexului. Axonii acestor celule, împreună cu fibrele târâtoare care trec prin stratul granular în tranzit și care transportă impulsuri de la măslinele inferioare către cerebel, ajung în stratul molecular superficial al cerebelului. Aici, axonii celulelor stratului granular și fibrele târâtoare se împart într-o formă de T, iar în stratul molecular ramurile lor iau o direcție longitudinală față de suprafața cerebelului. Impulsurile care au ajuns în stratul molecular al cortexului, trecând prin contactele sinaptice, cad pe dendritele ramificate ale celulelor Purkinje situate aici. Apoi urmăresc dendritele celulelor Purkinje până în corpurile lor, situate la granița straturilor moleculare și granulare. Apoi, de-a lungul axonilor acelorași celule care traversează stratul granular, ele pătrund în profunzimea substanței albe. Axonii celulelor Purkinje se termină în nucleii cerebelului. În principal în nucleul dintat. Impulsurile eferente care provin din cerebel de-a lungul axonilor celulelor care alcătuiesc nucleul acestuia și care participă la formarea pedunculilor cerebelosi părăsesc cerebelul.
Cerebelul are trei perechi de picioare: jos, mijloc și sus. Piciorul inferior îl conectează cu medula oblongata, mijlocul - cu puntea, cel superior - cu mezencefalul. Picioarele creierului formează căile care transportă impulsurile către și dinspre cerebel.
Vermisul cerebelos asigură stabilizarea centrului de greutate al corpului, echilibrul acestuia, stabilitatea, reglarea tonusului grupelor musculare reciproce, în principal a gâtului și a trunchiului, precum și apariția sinergiilor cerebeloase fiziologice care stabilizează echilibrul organismului.
Pentru a menține cu succes echilibrul corpului, cerebelul primește în mod constant informații care trec de-a lungul căilor spinocerebeloase de la proprioceptorii diferitelor părți ale corpului, precum și de la nucleii vestibulari, măsline inferioare, formațiunea reticulară și alte formațiuni implicate în controlul pozitia partilor corpului in spatiu. Majoritatea căilor aferente care duc la cerebel trec prin pediculul cerebelos inferior, unele dintre ele fiind situate în pediculul cerebelos superior.
Impulsuri de sensibilitate proprioceptiva, mergând spre cerebel, ca și alte impulsuri senzoriale, urmând dendritele primilor neuroni senzoriali, ajung în corpurile acestora situate în nodulii spinali. Ulterior, impulsurile care merg către cerebel de-a lungul axonilor acelorași neuroni sunt direcționate către corpurile neuronilor secunde, care sunt situate în părțile interioare ale bazei coarnelor posterioare, formând așa-numitul stâlpii lui Clark. Axonii lor cad în secțiunile laterale ale cordurilor laterale ale măduvei spinării, unde se formează căi spinocerebeloase, în acest caz, o parte a axonilor cade în coloana laterală a aceleiași părți și se formează acolo tractul spinocerebelos posterior al Flexig (tractus spinocerebelos posterior). O altă parte a axonilor celulelor coarnelor posterioare trece pe cealaltă parte a măduvei spinării și intră în cordonul lateral opus, formându-se în ea tractul spinocerebelos anterior al lui Govers (tractul spinocerebelos anterior). Tracturile spinocerebeloase, crescând în volum la nivelul fiecărui segment spinal, se ridică până la medular oblongata.
În medula oblongata, calea spinocerebeloasă posterioară deviază în direcția laterală și, după ce a trecut prin pediculul cerebelos inferior, pătrunde în cerebel. Calea spinocerebeloasă anterioară trece prin medula oblongata, puțul creierului, și ajunge la mezencefal, la nivelul căruia își face a doua intersecție în velul cerebral anterior și trece în cerebel prin pedunculul cerebelos superior.
Astfel, dintre cele două tracturi spinale, una nu este niciodată traversată (calea lui Fleksig neîncrucișată), iar cealaltă trece de două ori pe partea opusă (de două ori traversată de Govers). Ca urmare, ambii conduc impulsuri din fiecare jumătate a corpului, în principal către jumătatea homolaterală a cerebelului.
În plus față de tracturile spinocerebeloase ale lui Fleksig, impulsurile către cerebel trec prin pediculul cerebelos inferior de-a lungul tractul vestibulocerebelos (tractus vestibulocerebellaris), începând în principal în nucleul vestibular superior al spondilitei anchilozante și de-a lungul tractul olivomocerebelos (tractus olivocerebellaris) provenit din măslinul inferior. O parte din axonii celulelor nucleilor subțiri și în formă de pană, nu participă la formarea tractului bulbotalamic, sub formă de fibre arcuate externe (fibre arcuatae externae) intră şi în cerebel prin pedunculul cerebelos inferior.
Prin picioarele sale mijlocii, cerebelul primește impulsuri de la cortexul cerebral. Aceste impulsuri trec prin căi cortico-cerebelopontine, formate din doi neuroni. Corpurile primilor neuroni sunt localizate în cortexul cerebral, în principal în cortexul părților posterioare ale lobilor frontali. Axonii lor trec ca parte a coroanei radiante, piciorul anterior al capsulei interne și se termină în nucleii punții. Axonii celulelor neuronilor secunde, ale căror corpuri sunt localizate în propriile lor nuclee ale punții, mergi pe partea sa opusă și alcătuiește, după intersecție, pediculul cerebelos mijlociu,
care se termină în emisfera opusă a cerebelului.
Unele dintre impulsurile care au apărut în cortexul cerebral ajung în emisfera opusă a cerebelului, aducând informații nu despre mișcarea produsă, ci doar despre mișcarea activă planificată. Primind astfel de informații, cerebelul trimite instantaneu impulsuri care corectează mișcările voluntare, în principal, prin stingerea inerţiei şi cel mai rațional reglarea tonusului muscular reciproc - agonişti şi antagonişti musculari. Drept urmare, un fel de eimetrie, facand miscarile voluntare clare, perfectionate, lipsite de componente neadecvate.
Căile care părăsesc cerebelul sunt compuse din axonii celulelor, ale căror corpuri îi formează nucleele. Cele mai eferente căi, inclusiv căi de la nucleii dintați, lasa cerebelul prin piciorul superior. La nivelul tuberculilor inferiori ai cvadruplui se încrucișează tractul cerebelos eferent (intersecția picioarelor cerebeloase superioare ale lui Werneking). După ce a traversat fiecare dintre ele ajunge la nucleii roșii din partea opusă a mezencefalului. În nucleii roșii, impulsurile cerebeloase trec la următorul neuron și apoi se deplasează de-a lungul axonilor celulelor, ale căror corpuri sunt încorporate în nucleii roșii. Acești axoni se formează în căi roșu-spinală (tracti rubro spinalis), potecile lui Monakov, care la scurt timp după ieșirile din sâmburi roșii sunt supuse unei cruce (cruce de anvelope sau cruce de păstrăv), după care coboară în măduva spinării. În măduva spinării, căile spinării roșii-nucleare sunt situate în cordoanele laterale; fibrele lor constitutive se termină la celulele coarnelor anterioare ale măduvei spinării.
Întreaga cale eferentă de la cerebel la celulele coarnelor anterioare ale măduvei spinării poate fi numită cerebelos-rosu-nuclear-spinal (tractus cerebelo-rubrospinalis). El traversează de două ori (intersecția pedunculilor cerebelosi superiori și intersecția operculului) și în cele din urmă conectează fiecare emisferă cerebeloasă cu motoneuronii periferici localizați în coarnele anterioare ale jumătății homolaterale a măduvei spinării.
De la nucleii vermisului cerebelos, căile eferente trec în principal prin pediculul cerebelos inferior până la formarea reticulară a trunchiului cerebral și a nucleilor vestibulari. De aici, de-a lungul căilor reticulo-spinală și vestibulo-spinală care trec de-a lungul cordurilor anterioare ale măduvei spinării, ajung și la celulele coarnelor anterioare. O parte din impulsurile care provin din cerebel, trecând prin nucleii vestibulari, intră în fascicul longitudinal medial, ajunge la nucleii III, IV și VI ai nervilor cranieni care asigură mișcarea globilor oculari și afectează funcția acestora.
Pe scurt, trebuie subliniate următoarele:
1. Fiecare jumătate a cerebelului primește impulsuri în principal a) din jumătatea homolaterală a corpului, b) din emisfera opusă a creierului, care are conexiuni cortico-spinale cu aceeași jumătate a corpului.
(2) Din fiecare jumătate a cerebelului, impulsurile eferente sunt direcționate către celulele coarnelor anterioare ale jumătății homolaterale a măduvei spinării și către nucleii nervilor cranieni care asigură mișcarea globilor oculari.
Această natură a conexiunilor cerebeloase face posibilă înțelegerea de ce, atunci când o jumătate din cerebel este afectată, tulburările cerebeloase apar mai ales în același, adică. homolateral, jumătate din corp. Acest lucru este deosebit de pronunțat atunci când emisferele cerebeloase sunt afectate.
7.2. CERCETAREA FUNCȚIILOR CEREBELEI
ȘI MANIFESTĂRI CLINICE ALE ÎNCĂRGĂRILOR EI
Cu afectarea cerebelului, sunt caracteristice tulburări de statică și coordonare a mișcărilor, hipotonie musculară și nistagmus.
Leziune cerebeloasă pentru inceput viermele lui, duce la încălcări ale staticii - capacitatea de a menține o poziție stabilă a centrului de greutate al corpului uman, echilibru, stabilitate. Când această funcție este deranjată, ataxie statică (din greaca ataxia - tulburare, instabilitate). Se remarcă instabilitatea pacientului. Prin urmare, în poziție în picioare, își desfășoară picioarele larg, se echilibrează cu mâinile. Ataxia statică în mod deosebit este detectată cu o scădere artificială a zonei de sprijin, în special în ipostaza Romberg. Pacientul este invitat să se ridice, mișcându-și ferm picioarele și ridicând ușor capul. În prezența tulburărilor cerebeloase, se remarcă instabilitatea pacientului în această poziție, corpul lui se balansează, uneori „trage” într-o anumită direcție, iar dacă pacientul nu este susținut, poate cădea. În cazul deteriorării viermelui cerebelos, pacientul se leagănă de obicei dintr-o parte în alta și adesea cade înapoi. Cu patologia emisferei cerebeloase, există o tendință de a cădea în principal spre focalizarea patologică. Dacă tulburarea statică este moderat exprimată, este mai ușor de identificat în așa-numita complicat sau poza Romberg sensibilizată. Pacientul este rugat să-și pună picioarele într-o linie, astfel încât degetul unui picior să se sprijine pe călcâiul celuilalt. Evaluarea stabilității este aceeași ca în poziția obișnuită a Romberg.
În mod normal, atunci când o persoană stă în picioare, mușchii picioarelor sale sunt încordați. (reacție de sprijin), cu amenințarea de a cădea în lateral, piciorul său de pe această parte se mișcă în aceeași direcție, iar celălalt picior iese de pe podea (reacție la salt). Cu afectarea cerebelului (în principal viermele), reacțiile pacientului sunt perturbate
sprijin si sari. Încălcarea reacției de sprijin se manifestă prin instabilitatea pacientului în poziția în picioare, în special în poziția Romberg. Încălcarea reacției de săritură duce la faptul că, dacă medicul, stând în spatele pacientului și asigurându-l, împinge pacientul într-o direcție sau alta, atunci pacientul cade cu o ușoară împingere (simptomul de împingere).
Cu afectarea cerebelului, mersul pacientului este de obicei schimbat din cauza dezvoltării ataxie statolocomotorie. Mersul cerebelos în multe privințe seamănă cu mersul unui beat, de aceea este uneori numit „mersul unui beat”. Din cauza instabilității, pacientul merge nesigur, desfăcându-și picioarele larg depărtate, în timp ce este „aruncat” dintr-o parte în alta. Iar atunci când emisfera cerebeloasă este deteriorată, se abate la mers dintr-o direcție dată către focarul patologic. Instabilitatea este deosebit de pronunțată la viraj. Dacă ataxia este pronunțată, atunci pacienții își pierd complet capacitatea de a-și controla corpul și nu pot doar să stea și să meargă, ci chiar să se așeze.
Leziunea predominantă a emisferelor cerebeloase duce la o tulburare a efectelor sale antiinerțiale, în special la apariția ataxie cinetică. Se manifestă prin stângăcia mișcărilor și se manifestă mai ales la mișcările care necesită precizie. Pentru detectarea ataxiei cinetice se efectuează teste de coordonare a mișcărilor. Unele dintre ele sunt descrise mai jos.
Test pentru diadococineză (din greacă. diadochos – secvență). Pacientul este invitat să închidă ochii, să-și întindă brațele înainte și rapid, să supineze ritmic și să pătrundă în mâini. În caz de deteriorare a emisferei cerebeloase, mișcările mâinii pe partea procesului patologic se dovedesc a fi mai ample (o consecință a dismetriei, mai precis, a hipermetriei), ca urmare, mâna începe să rămână în urmă. Aceasta indică prezența adiadocokinezei.
Testul degetelor. Un pacient cu ochii închiși trebuie să-și retragă mâna și apoi, încet, cu degetul arătător, să atingă vârful nasului. În cazul patologiei cerebeloase, mâna din partea focarului patologic face o mișcare excesivă în volum (hipermetrie), drept urmare pacientul ratează. Un test deget-nas dezvăluie o caracteristică a patologiei cerebeloase tremor cerebelos (intenționat), a cărei amplitudine crește pe măsură ce degetul se apropie de țintă. Acest test vă permite să identificați așa-numita braditelechinezie. (simptomul de căpăstru): nu departe de țintă, mișcarea degetului încetinește, uneori chiar se oprește și apoi se reia.
Test deget-deget. Un pacient cu ochii închiși este invitat să-și întindă larg brațele și apoi să apropie degetele arătător, încercând să bage degetul în deget, în timp ce, ca și în cazul testului cu degetul, se dezvăluie un tremor intenționat și un simptom de căpăstru.
Testul genunchiului calcanean (fig. 7.3). Pacientul, întins pe spate cu ochii închiși, este rugat să ridice un picior sus și apoi să lovească genunchiul celuilalt picior cu călcâiul. Cu patologia cerebeloasă, pacientul nu poate sau îi este greu să-și bage călcâiul în genunchiul celuilalt picior, mai ales când se efectuează un test cu piciorul homolateral de emisfera cerebeloasă afectată. Dacă, totuși, călcâiul ajunge până la genunchi, atunci se propune să-l țină, atingând ușor suprafața frontală a piciorului inferior, până la articulația gleznei, în timp ce în cazul patologiei cerebeloase, călcâiul alunecă de pe picior. timpul într-o direcție sau alta.

Orez. 7.3.Testul genunchiului calcanean.
Test orientativ: Pacientul este invitat să lovească de câteva ori vârful de cauciuc al ciocanului, care se află în mâna examinatorului, cu degetul arătător. În cazul patologiei cerebeloase în mâna pacientului pe partea laterală a emisferei cerebeloase afectate, există o nealiniere din cauza dismetriei.
Simptomul Tom-Jumenti: Dacă pacientul ridică un obiect, cum ar fi un pahar, își desfășoară degetele excesiv.
Nistagmus cerebelos. Convulsii ale globilor oculari atunci când priviți în lateral (nistagmus orizontal) este considerată o consecință a tremurului intenționat al globilor oculari (vezi capitolul 30).
Tulburare de vorbire: Vorbirea își pierde fluiditatea, devine explozivă, fragmentată, scanată ca disartria cerebeloasă (vezi capitolul 25).
Schimbarea scrisului de mână: Din cauza dereglării în coordonarea mișcărilor mâinii, scrisul devine neuniform, literele sunt deformate, excesiv de mari (megalografie).
Fenomen pronator: Pacientului i se cere să-și țină brațele întinse în poziția de supinație, în timp ce pronația spontană apare curând pe partea emisferei cerebeloase afectate.
Simptomul Hoff-Schilder: Dacă pacientul își ține brațele întinse înainte, atunci pe partea emisferei afectate, brațul este în curând retras spre exterior.
Un fenomen de imitație. Un pacient cu ochii închiși ar trebui să dea rapid mâinii o poziție similară cu cea pe care examinatorul o acordase anterior celeilalte mâini. Când emisfera cerebeloasă este deteriorată, mâna homolaterală face o mișcare cu amplitudine excesivă.
fenomenul lui Doinikov. Fenomenul degetelor. Pacientul așezat este invitat să-și pună mâinile supinate cu degetele depărtate pe coapse și să închidă ochii. În cazul unei leziuni a cerebelului pe partea focarului patologic, în curând apar flexia spontană a degetelor și pronația mâinii și antebrațului.
Simptomul Stuart-Holmes. Examinatorul îi cere pacientului așezat pe scaun să îndoaie antebrațele supinate și în același timp, luându-i mâinile de încheieturi, îi rezistă. Dacă în același timp eliberați brusc mâinile pacientului, atunci mâna de pe partea afectată, îndoită prin inerție, îl va lovi cu forță în piept.
Hipotensiunea musculară. Înfrângerea vermisului cerebelos duce de obicei la hipotensiune musculară difuză. Odată cu înfrângerea emisferei cerebeloase, mișcările pasive relevă o scădere a tonusului muscular pe partea procesului patologic. Hipotonia musculară duce la posibilitatea de hiperextensie a antebrațului și a piciorului inferior (Simptomul Olshansky) cu miscari pasive, la aparenta simptome ale unei mâini sau picior atârnate cu tremuratul lor pasiv.
Asinergii cerebeloase patologice. Încălcări ale sinergiilor fiziologice în timpul actelor motorii complexe sunt relevate, în special, în timpul următoarelor teste (Fig. 7.4).
1. Asinergie după Babinsky în poziție în picioare. Dacă un pacient care stă în picioare cu picioarele deplasate încearcă să se aplece înapoi, aruncându-și capul pe spate, atunci în mod normal, în acest caz, are loc flexia articulațiilor genunchiului. În patologia cerebeloasă din cauza asinergiei, această mișcare prietenoasă este absentă, iar pacientul, pierzându-și echilibrul, cade înapoi.
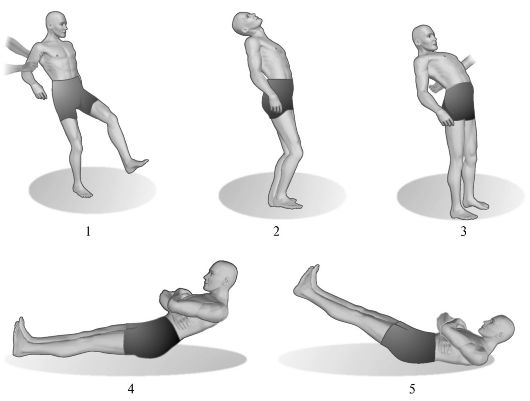
Orez. 7.4.Asinergie cerebeloasă.
1 - mersul unui pacient cu ataxie cerebeloasă severă; 2 - înclinarea spate a corpului este normală; 3 - cu afectarea cerebelului, pacientul, aplecându-se înapoi, nu poate menține echilibrul; 4 - efectuarea unui test pentru asinergia cerebeloasă conform lui Babinsky de către o persoană sănătoasă; 5 - efectuarea aceluiaşi test la pacienţii cu leziuni cerebeloase.
2. Asinergie după Babinsky în decubit dorsal. Pacientul, întins pe un plan ferm, cu picioarele întinse, întinse pe lățimea centurii umărului, este invitat să-și încrucișeze brațele peste piept și apoi să se așeze. În prezența patologiei cerebeloase din cauza absenței contracției prietenoase a mușchilor fesieri (manifestarea asinergiei), pacientul nu poate fixa picioarele și pelvisul pe zona de sprijin, ca urmare, picioarele se ridică și nu se poate așeza. Semnificația acestui simptom nu trebuie supraestimată la pacienții vârstnici, la persoanele cu peretele abdominal flasc sau obez.
Rezumând cele de mai sus, trebuie subliniată diversitatea și importanța funcțiilor îndeplinite de cerebel. Ca parte a unui mecanism complex de reglare a feedback-ului, cerebelul acționează ca un punct focal pentru echilibrarea corpului și menținerea tonusului muscular. După cum notează P. Duus (1995), cerebelul oferă capacitatea de a efectua mișcări discrete și precise, autorul crede în mod rezonabil că cerebelul funcționează ca un computer, urmărind și coordonând informațiile senzoriale la intrare și simulând semnalele motorii la ieșire.
7.3. DEGENERAȚII MULTISISTEME
Cu semne de patologie cerebeloasă
Degenerările multisistemice reprezintă un grup de boli neurodegenerative, a căror caracteristică comună este natura multifocală a leziunii cu implicarea diferitelor sisteme funcționale și neurotransmițătoare ale creierului în procesul patologic și, prin urmare, natura polisistemică a manifestărilor clinice.
7.3.1. Ataxie cerebeloasă
Ataxiile spinocerebeloase includ boli degenerative ereditare progresive, în care sunt afectate în principal structurile cerebelului, trunchiului cerebral și căile măduvei spinării, care sunt în principal legate de sistemul extrapiramidal.
7.3.1.1. ataxia ereditară a lui Friedreich
Boală ereditară descrisă în 1861 de neuropatologul german N. Friedreich (Friedreich N., 1825-1882). Se moștenește într-o manieră autozomal recesivă sau (mai rar) într-o manieră autosomal dominantă cu penetranță incompletă și expresie variabilă a genei. Sunt posibile și cazuri sporadice de boală.
Patogenezaboala nespecificata. În special, nu există nicio idee despre defectul biochimic primar care constituie baza acestuia.
Patomorfologie.Studiile patologice relevă o subțiere pronunțată a măduvei spinării din cauza proceselor atrofice din cordoanele posterioare și laterale ale acesteia. De regulă, căile în formă de pană (Burdach) și blând (Gaulle) și căile spinocerebeloase ale lui Govers și Fleksig suferă, precum și calea piramidală încrucișată care conține
multe fibre legate de sistemul extrapiramidal. Procesele degenerative se exprima si in cerebel, in substanta sa alba si in aparatul nuclear.
Manifestari clinice. Boala se manifestă la copii sau tineri sub 25 de ani. S.N. Davidenkov (1880-1961) a remarcat că mai des semnele clinice ale bolii apar la copiii cu vârsta de 6-10 ani. Primul semn de boală este de obicei ataxia. Pacienții se confruntă cu incertitudine, eșalonări la mers, modificări ale mersului (la mers, picioarele sunt larg depărtate). Mersul în boala Friedreich poate fi numit tabetic-cerebelos, deoarece modificările sale sunt cauzate de o combinație de ataxie sensibilă și cerebeloasă, precum și de o scădere pronunțată de obicei a tonusului muscular. De asemenea, sunt caracteristice tulburările de statică, dezordonarea mâinilor, tremor intenționat, disartria. Posibil nistagmus, pierderea auzului, elemente de intonare a vorbirii, semne de insuficiență piramidală (hiperreflexie tendinoasă, reflexe patologice ale picioarelor, uneori o ușoară creștere a tonusului muscular), impuls imperativ de a urina, scăderea potenței sexuale. Uneori apare hiperkineza atetoidă.
O tulburare precoce de sensibilitate profundă duce la o scădere progresivă a reflexelor tendinoase: mai întâi pe picioare, apoi pe brațe. În timp, se formează hipotrofia musculară a picioarelor distale. Prezența anomaliilor în dezvoltarea scheletului este caracteristică. În primul rând, acest lucru se manifestă prin prezență Picioarele lui Friedreich: piciorul este scurtat, „gol”, cu un arc foarte înalt. Falangele principale ale degetelor ei sunt neîndoite, restul sunt îndoite (Fig. 7.5). Posibilă deformare a coloanei vertebrale, a pieptului. Există adesea manifestări de cardiopatie. Boala progresează lent, dar în mod constant duce la invaliditatea pacienților care în cele din urmă devin imobilizați la pat.
Tratament. Tratamentul patogenetic nu a fost dezvoltat. Prescripționați medicamente care îmbunătățesc metabolismul în structurile sistemului nervos, agenți de fortificare. Cu deformarea severă a picioarelor, sunt indicați încălțămintea ortopedică.
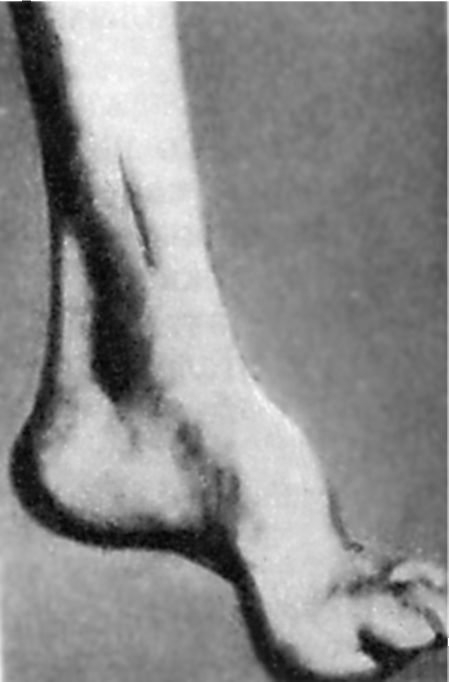
Orez. 7.5.piciorul lui Friedreich.
7.3.1.2. Ataxie cerebeloasă ereditară (boala lui Pierre Marie)
Aceasta este o boală ereditară cronică progresivă, manifestată la vârsta de 30-45 de ani, cu tulburări cerebeloase cu creștere lentă în combinație cu semne de insuficiență piramidală, în timp ce sunt caracteristice ataxia cerebeloasă statică și dinamică, tremurăturile intenționate, vorbirea scanată, hiperreflexia tendonului. Posibile clonuri, reflexe piramidale patologice, strabism, scăderea vederii, îngustarea câmpurilor vizuale din cauza atrofiei primare a nervilor optici și a degenerării pigmentare a retinei. Cursul bolii este lent progresiv. Există o scădere a dimensiunii cerebelului, degenerarea celulară
Purkinje, măsline inferioare, tracturi spinocerebeloase. Se moștenește în mod autosomal dominant. Boala a fost descrisă în 1893 de neuropatologul francez R. Marie (1853-1940).
În prezent, nu există unanimitate în înțelegerea termenului „boala lui Pierre Marie”, iar întrebarea posibilității de a-l separa într-o formă nosologică independentă este discutabilă.
Nu a fost dezvoltat niciun tratament. De obicei, se folosesc agenți metabolic activi și restauratori, precum și simptomatici.
7.3.2. Distrofia olivopontocerebeloasă (boala Dejerine-Thom)
Acesta este un grup de boli ereditare cronice progresive, în care modificările distrofice se dezvoltă în principal la nivelul cerebelului, măslinelor inferioare, în nucleii proprii ai puțului și în structurile creierului asociate acestora.
Odată cu dezvoltarea bolii la o vârstă fragedă, aproximativ jumătate din cazuri sunt moștenite în mod dominant sau recesiv, restul sunt sporadice. În cazurile sporadice de boală, manifestările sindromului akinetic-rigid și insuficiența autonomă progresivă sunt mai frecvente. Vârsta medie a pacientului cu manifestarea formei ereditare a bolii în fenotip este de 28 de ani, cu sporadic - 49 de ani, speranța medie de viață este de 14,9, respectiv 6,3 ani. În forma sporadică, pe lângă atrofia măslinelor, pontului și cerebelului, se găsesc mai des leziuni ale cordoanelor laterale ale măduvei spinării, substanței negre și striatum, o pată albăstruie în fosa romboidă a ventriculului al patrulea al creierului. .
Simptomele sindromului cerebelos în creștere sunt caracteristice. Sunt posibile tulburări de sensibilitate, elemente de sindroame bulbare și akinetic-rigide, hiperkinezie, în special miorritmii la nivelul limbii și palatului moale, oftalmopareza, scăderea acuității vizuale, tulburări intelectuale. Boala a fost descrisă în 1900 de neuropatologii francezi J. Dejerine și A. Thomas.
Boala debutează adesea cu tulburări de mers - sunt posibile instabilitate, dezordonare, căderi neașteptate. Aceste tulburări pot fi singura manifestare a bolii în decurs de 1-2 ani. În viitor, apar și cresc tulburări de coordonare la nivelul mâinilor: manipulările cu obiecte mici sunt dificile, scrisul de mână este perturbat, apare un tremur intenționat. Vorbirea devine intermitentă, neclară, cu o tentă nazală și ritmul respirației care nu corespunde structurii vorbirii (pacientul vorbește ca și cum ar fi fost sugrumat). În acest stadiu al bolii se unesc manifestările de insuficiență autonomă progresivă, apar semne de sindrom akinetic-rigid. Uneori simptomele dominante pentru pacient sunt disfagia, atacurile de sufocare nocturnă. Se dezvoltă în legătură cu pareza mixtă a mușchilor bulbari și pot pune viața în pericol.
În 1970, neuropatologii germani B.W. Konigsmark și L.P. Weiner a remarcat 5 tipuri principale distrofia olivopontocerebeloasă, diferită fie prin manifestări clinice și morfologice, fie prin tipul de moștenire.
eu tip (tip Menzel). La vârsta de 14-70 (mai des 30-40) ani, se manifestă ataxie, disartrie, disfonie, hipotonie musculară, în stadiul târziu - un tremur gros al capului, trunchiului, brațelor, mușchilor, semne de acinetic- sindrom rigid. Posibile semne piramidale patologice, pareza privirii, oftalmoplegie externa si interna, tulburari de sensibilitate, dementa. Se moștenește în mod autosomal dominant. A fost evidențiată ca formă independentă în 1891 de către P. Menzel.
II tip (tipul Fickler-Winkler) ... La varsta de 20-80 de ani se manifesta ataxie, scaderea tonusului muscular si reflexe tendinoase. Se moștenește în mod autosomal recesiv. Sunt posibile cazuri sporadice.
III tip cu degenerare retiniană. Se manifestă în copilărie sau în vârstă tânără (până la 35 de ani), ataxie, tremor la nivelul capului și extremităților, disartrie, semne de insuficiență piramidală, scăderea progresivă a vederii cu un rezultat în orbire; posibil nistagmus, oftalmoplegie, uneori tulburări senzoriale disociate. Se moștenește în mod autosomal dominant.
IV tip (tipul Jester-Highmaker). La vârsta de 17-30 de ani, debutează cu ataxie cerebeloasă sau semne de parapareză spastică inferioară, în ambele cazuri, deja într-un stadiu incipient al bolii, se formează o combinație a acestor manifestări, la care elemente de sindrom bulbar, pareză. a mușchilor faciali și se adaugă ulterior tulburări de sensibilitate profundă. Dominant moștenit.
V tip de. Se manifestă la vârsta de 7-45 ani sunt posibile ataxie, disartrie, semne de sindrom akinetic-rigid și alte tulburări extrapiramidale, oftalmoplegie progresivă și demență. Dominant moștenit.
7.3.3. Degenerescenta olivorubrocerebeloasa (sindrom Lejeune-Lermitte, boala Lermitte)
Boala se caracterizează prin atrofia progresivă a cerebelului, în principal a cortexului său, a nucleilor dințați și a pedunculilor cerebelosi superiori, a măslinelor inferioare și a nucleilor roșii. Se manifestă în primul rând prin ataxie statică și dinamică; în viitor, sunt posibile alte semne ale sindromului cerebelos și afectarea trunchiului cerebral. Boala a fost descrisă de neuropatologii francezi J. Lhermitte (Lhermitte J.J., 1877-1959) și J. Lezhon (Lejonne J., născut în 1894).
7.3.4. Atrofie multisistem
În ultimele decenii, o boală neurodegenerativă sporadică, progresivă, numită atrofie multisistem, a fost identificată ca o formă independentă. Se caracterizează printr-o leziune combinată a ganglionilor bazali, cerebelului, trunchiului cerebral, măduvei spinării. Principalele manifestări clinice: parkinsonism, ataxie cerebeloasă, semne de insuficiență piramidală și autonomă (Levin O.S., 2002). În funcție de predominanța anumitor caracteristici ale tabloului clinic, se disting trei tipuri de atrofie multisistem.
1) tip olivopontocerebelos, caracterizat prin predominarea semnelor de atac cerebelos;
2) tip strionigral, la care domină semnele de parkinsonism;
3) Sindromul Shai-Drager, caracterizat prin predominarea în tabloul clinic a semnelor de insuficiență autonomă progresivă cu simptome de hipotensiune arterială ortostatică.
Baza atrofiei multisistemice este degenerarea selectivă a anumitor zone ale substanței predominant cenușii a creierului cu afectarea neuronilor și a elementelor gliale. Cauzele manifestărilor degenerative în țesutul cerebral rămân necunoscute astăzi. Manifestările atrofiei multisistemice de tip olivopontocerebelos sunt asociate cu afectarea celulelor Purkinje din cortexul cerebelos, precum și a neuronilor măslinelor inferioare, nucleilor pontocerebelos, demielinizării și degenerarii, în principal a căilor pontocerebeloase.
Tulburările cerebeloase sunt de obicei ataxie statică și dinamică cu mișcare locomotorie afectată. Caracterizat prin instabilitate în poziția Romberg, ataxie la mers, dismetrie, adiadococineză, tremor intenționat, poate exista nistagmus (vertical orizontal, bătând în jos), mișcări intermitente și lente de urmărire a privirii, convergență afectată a ochilor, vorbire scanată.
Atrofia multisistemică apare de obicei la vârsta adultă și progresează rapid. Diagnosticul se bazează pe dovezi clinice și se caracterizează printr-o combinație de semne de parkinsonism, insuficiență cerebeloasă și tulburări autonome. Tratamentul bolii nu a fost dezvoltat. Durata bolii - în 10 ani, se termină cu moartea.
7.4. ALTE BOLI ASOCIATE CU SIMPTOME DE BOLI CEREBRALE
Dacă pacientul prezintă semne de leziune cerebeloasă, atunci în majoritatea cazurilor, în primul rând trebuie să te gândești la posibilitate tumori cerebeloase(astrocitom, angioblastom, meduloblastom, tumori metastatice) sau scleroza multiplă. La tumori cerebeloase apar semne precoce ale hipertensiunii intracraniene. În scleroza multiplă, de obicei, este posibil să se identifice, pe lângă patologia cerebeloasă, manifestări clinice de afectare a altor structuri ale sistemului nervos central, în primul rând sistemul vizual și piramidal. În neurologia clasică, caracteristica a scleroză multiplă Triada lui Charcot: nistagmus, tremor intenționat și vorbire scanată și Sindromul Nonne: tulburări de coordonare a mișcărilor, dismetrie, vorbire cântată și asinergie cerebeloasă.
Tulburările cerebeloase sunt majore și în sindromul Mann posttraumatic, care se caracterizează prin ataxie, discoordonare, asinergie, nistagmus. Trauma sau infecția poate provoca cerebelos Sindromul Goldstein-Reichmann: tulburări de statică și coordonare a mișcărilor, asinergie, tremor intenționat, scăderea tonusului muscular, hipermetrie, megalografie, percepție afectată a masei (greutății) unui obiect în mâini.
Tulburările funcției cerebeloase pot fi, de asemenea, de natură congenitală, manifestându-se, în special, Sindromul Zeeman: ataxie, întârzierea dezvoltării vorbirii și, ulterior, disartrie cerebeloasă.
Ataxie cerebeloasă congenitală Se manifestă printr-o întârziere a dezvoltării funcțiilor motorii ale copilului (la vârsta de 6 luni nu poate sta, începe să meargă târziu, în timp ce mersul este atactic), precum și întârzierea vorbirii, conservarea prelungită a disartriei, uneori psihică. retardare, iar manifestările microcraniene nu sunt neobișnuite. La CT, emisferele cerebeloase sunt reduse. Până la vârsta de aproximativ 10 ani, apare de obicei compensarea funcțiilor creierului, care, totuși, poate fi perturbată sub influența influențelor exogene dăunătoare. Sunt posibile și forme progresive ale bolii.
O manifestare a hipoplaziei congenitale a cerebelului este și Sindromul Fancony-Turner. Se caracterizează prin tulburări de statică și coordonare a mișcărilor, nistagmus, care sunt de obicei însoțite de retard mintal.
Congenital include, de asemenea, un tip moștenit autosomal recesiv, care este rar întâlnit boala lui Betten: Se caracterizează prin ataxie cerebeloasă congenitală, manifestată în primul an de viață prin tulburări de statică și coordonare a mișcărilor, nistagmus, tulburare de coordonare a privirii și hipotonie musculară moderată. Sunt posibile semne displazice. Copilul întârzie, uneori abia la 2-3 ani, începe să-și țină capul, chiar mai târziu - să stea, să meargă, să vorbească. Vorbirea lui este schimbată în funcție de tipul de disartrie cerebeloasă. Sunt posibile tulburări vegetativ-viscerale, manifestări de imunosupresie. După câțiva ani, tabloul clinic se stabilizează de obicei, pacientul se adaptează într-o oarecare măsură la defectele existente.
Ataxie spastică la sugestia lui A. Bell și E. Carmichel (1939), a fost numită ataxia cerebeloasă moștenită de tip autosomal dominant, care se caracterizează prin debutul bolii la vârsta de 3-4 ani și se manifestă printr-o combinație de cerebeloase. ataxie cu disartrie, hiperreflexie tendinoasă și tonus muscular crescut de tip spastic, în timp ce este posibilă (dar nu semne obligatorii ale bolii) atrofie a nervilor optici, degenerare retiniană, nistagmus, tulburări oculomotorii.
Autosomal dominant este moștenit sindromul Feldman(descris de medicul german H. Feldmann, născut în 1919): ataxie cerebeloasă, tremor intenționat și albire precoce a părului. Se manifestă în a doua decadă de viață și progresează în continuare încet, ducând la invaliditate după 20-30 de ani.
Atrofie cerebeloasă tardivă sau Sindromul lui Tom descrisă în 1906 de neurologul francez A. Thomas (1867-1963), se manifestă de obicei la persoanele peste 50 de ani cu atrofie progresivă a cortexului cerebelos. În fenotip apar semne ale sindromului cerebelos, în primul rând ataxie statică și locomotorie cerebeloasă, vorbire scanată și modificări ale scrisului de mână. Într-un stadiu mult avansat, sunt posibile manifestări de insuficiență piramidală.
Combinația tulburărilor cerebeloase cu mioclonia se caracterizează prin Hunt disinergia cerebeloasă mioclonică, sau ataxie mioclonică, cu acest complex de simptome în tabloul clinic se manifestă tremor intenționat, mioclon care apare în mâini, iar ulterior dobândind un caracter generalizat, se manifestă ataxie și disinergie, nistagmus, vorbire scanată, scăderea tonusului muscular. Este o consecință a degenerării nucleilor cerebelosi, a nucleilor roșii și a conexiunilor acestora, precum și a structurilor cortico-subcorticale.
Într-un stadiu avansat al bolii, sunt posibile crize epileptice și demență. Prognosticul este prost. Se referă la o formă rară de ataxie ereditară progresivă. Se moștenește în mod autosomal recesiv. De obicei apare la o vârstă fragedă. Independența nosologică a complexului de simptome este contestată. Neurologul american R. Hunt (1872-1937) a descris boala în 1921.
Printre procesele degenerative, un anumit loc este ocupat de degenerescenta cerebeloasa Holmes, sau atrofie cerebeloasă familială, sau atrofia progresivă a sistemului cerebelos, în principal a nucleilor dinţaţi, precum şi a nucleilor roşii, în timp ce manifestările de demielinizare sunt exprimate în pediculul cerebelos superior. Caracterizat prin ataxie statică și dinamică, asinergie, nistagmus, disartrie, scăderea tonusului muscular, distonie musculară, tremor capului, mioclonie. Crizele de epilepsie apar aproape simultan. Inteligența este de obicei păstrată. EEG arată aritmie paroxistică. Boala este recunoscută ca ereditară, dar nu este specificat tipul de moștenire. Descrisă boala în 1907 de către neuropatologul englez G. Holmes
(1876-1965).
Degenerescenta cerebeloasa alcoolica - o consecință a intoxicației alcoolice cronice. În principal este afectat viermele cerebelos, cu ataxie cerebeloasă și tulburări de coordonare a mișcărilor picioarelor manifestându-se în primul rând, în timp ce mișcările mâinii, funcțiile oculomotorii și de vorbire sunt afectate într-o măsură mult mai mică. De obicei, această boală este însoțită de o pierdere pronunțată a memoriei în combinație cu polineuropatia.
se manifestă ca ataxie cerebeloasă, care poate fi uneori singurul simptom clinic asociat unei tumori maligne, fără semne locale care să indice locul apariției acesteia. Degenerescenta cerebeloasa paraneoplazica poate fi, în special, o manifestare secundară a cancerului de sân sau ovarian.
Sindromul Barraquer-Bordas-Ruiz-Lara se manifestă ca tulburări cerebeloase care apar în legătură cu atrofia rapid progresivă a cerebelului. Un sindrom la pacienţii cu cancer bronşic însoţit de intoxicaţie generală este descris de medicul spaniol modern L. Barraquer-Bordas (născut în 1923).
Rareori găsit ataxie cromozomială X recesivă- o boală ereditară care se manifestă aproape doar la bărbații cu insuficiență cerebeloasă lent progresivă. Se transmite într-un tip recesiv, legat de sex.
De remarcat și ataxie paroxistica familială, sau ataxie periodică. Debutează mai des în copilărie, dar poate apărea și mai târziu - până la 60 de ani. Tabloul clinic se reduce la manifestări paroxistice de nistagmus, disartrie și ataxie, scăderea tonusului muscular, amețeli, greață, vărsături, cefalee, care durează de la câteva minute până la 4 săptămâni.
Atacurile de ataxie paroxistică familială pot fi declanșate de stres emoțional, oboseală fizică, febră, consum de alcool, în timp ce între atacuri simptomele neurologice focale în majoritatea cazurilor nu sunt depistate, dar uneori sunt posibile nistagmus și simptome cerebeloase ușoare.
Substratul morfologic al bolii este recunoscut ca un proces atrofic în principal în partea anterioară a viermelui cerebelos. Boala a fost descrisă pentru prima dată în 1946 de M. Parker. Se moștenește în mod autosomal dominant. În 1987, cu ataxie paroxistică familială, s-a constatat o scădere a activității piruvat dehidrogenazei leucocitelor din sânge până la 50-60% din nivelul normal. În 1977 R. Lafrance et al. a atras atenția asupra efectului profilactic ridicat al diacarbului, ulterior a fost propusă flunarizina pentru tratamentul ataxiei paroxistice familiale.
Ataxie cerebeloasă acută sau sindromul Leiden-Westphal, este un complex de simptome bine definit, care este o complicație parainfectioasă. Apare mai des la copii la 1-2 săptămâni după o infecție generală (gripă, tifos, salmoneloză etc.). Caracterizat prin ataxie statică și dinamică masivă, tremor intenționat, hipermetrie, asinergie, nistagmus, vorbire scanată, scăderea tonusului muscular. În lichidul cefalorahidian este detectată pleocitoza limfocitară, o creștere moderată a proteinei. La debutul bolii, sunt posibile amețeli, tulburări de conștiență, convulsii. Pe CT și RMN, patologia nu este detectată. Cursul este benign. În majoritatea cazurilor, după câteva săptămâni sau luni - recuperare completă, uneori - tulburări reziduale sub formă de insuficiență cerebeloasă ușoară.
boala Marie-Foix-Alajuanin - atrofie corticală simetrică tardivă a cerebelului cu o leziune predominantă a neuronilor piriformi (celule Purkinje) și a stratului granular al cortexului, precum și a părții bucale a vermisului cerebelos și degenerarea măslinelor. Se manifestă la persoanele de 40-75 de ani cu tulburări de echilibru, ataxie, tulburări de mers, tulburări de coordonare și scăderea tonusului muscular, în principal la nivelul picioarelor; tremorul intenționat în mâini nu este foarte pronunțat. Tulburările de vorbire sunt posibile, dar nu aparțin semnelor obligatorii ale bolii. Boala a fost descrisă în 1922 de neuropatologii francezi P. Marie, Ch. Foix și Th. Alajouanine. Boala este sporadica. Etiologia bolii nu a fost clarificată. Există opinii despre rolul provocator al intoxicației, în primul rând abuzul de alcool, precum și hipoxie, povara ereditară. Tabloul clinic este confirmat de datele CT ale capului, care relevă o scădere pronunțată a volumului cerebelului pe fondul proceselor atrofice difuze în creier. În plus, un nivel ridicat de aminotransferaze în plasma sanguină este recunoscut ca caracteristic (Ponomareva E.N. et al., 1997).
