7.1. STRUCTURE, CONNECTIONS AND FUNCTIONS OF THE CEREBEL
The cerebellum (cerebellum) is located under a duplicate dura mater known as outline of the cerebellum(tentorium cerebelli), which divides the cranial cavity into two unequal spaces - supratentorial and subtentorial. V subtentorial space, the bottom of which is the posterior cranial fossa, in addition to the cerebellum, is the brain stem. The volume of the cerebellum averages 162 cm 3. Its weight varies between 136-169 g.
The cerebellum is located above the bridge and the medulla oblongata. Together with the superior and inferior cerebral sails, it constitutes the roof of the fourth ventricle of the brain, the bottom of which is the so-called rhomboid fossa (see Chapter 9). Above the cerebellum are the occipital lobes of the large brain, separated from it by the tentorium of the cerebellum.
In the cerebellum, there are two hemispheres(hemispherum cerebelli). Between them, in the sagittal plane above the IV ventricle of the brain, the phylogenetically most ancient part of the cerebellum is located - its worm(vermis cerebelli). The vermis and cerebellar hemispheres are fragmented into lobules by deep transverse grooves.
The cerebellum is composed of gray and white matter. The gray matter forms the cerebellar cortex and the paired nuclei nuclei cerebelli located in its depth (Fig. 7.1). The largest of them are jagged kernels(nucleus dentatus) - located in the hemispheres. In the central part of the worm there are tent cores(nuclei
Rice. 7.1. Cerebellar nuclei.
1 - toothed core; 2 - corky core; 3 - the core of the tent; 4 - spherical nucleus.
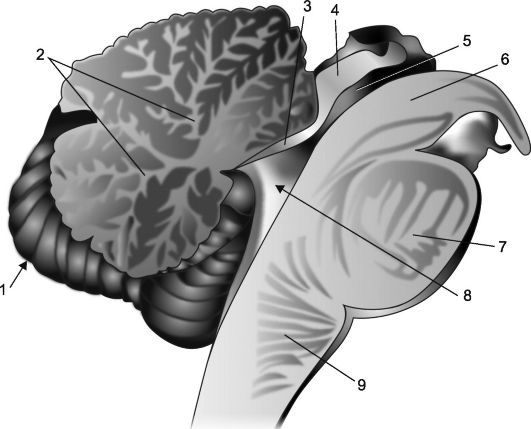
Rice. 7.2.Sagittal section of the cerebellum and brainstem.
1 - cerebellum; 2 - "tree of life"; 3 - forebrain sail; 4 - plate of the quadruple; 5 - aqueduct of the brain; 6 - the leg of the brain; 7 - bridge; 8 - IV ventricle, its choroid plexus and tent; 9 - medulla oblongata.
fastigii), between them and the dentate nuclei are spherical and corky nuclei(nuctei.globosus et emboliformis).
Due to the fact that the cortex covers the entire surface of the cerebellum and penetrates into the depths of its furrows, on a sagittal section of the cerebellum, its tissue has a leaf pattern, the veins of which are formed by a white matter (Fig. 7.2), which makes up the so-called tree of life of the cerebellum (arbor vitae cerebelli). At the base of the tree of life there is a wedge-shaped notch, which is the upper part of the cavity of the IV ventricle; the edges of this recess form his tent. The roof of the tent is the cerebellar worm, and its anterior and posterior walls are thin cerebral plates known as the anterior and posterior brain sails(vella medullare anterior et posterior).
Some information about cerebellar architectonics, giving grounds for judging the function of its components. Have cerebellar cortex There are two cell layers: the inner one is granular, consisting of small grain cells, and the outer one is molecular. Between them there is a number of large pear-shaped cells bearing the name of the Czech scientist I. Purkinje who described them (Purkinje I., 1787-1869).
The impulses enter the cerebellar cortex through mossy and creeping fibers penetrating into it from the white matter, which make up the afferent pathways of the cerebellum. Through mossy fibers, impulses from the spinal cord
vestibular nuclei and nuclei of the pons are transferred to the cells of the granular layer of the cortex. The axons of these cells, together with creeping fibers passing through the granular layer in transit and carrying impulses from the inferior olives to the cerebellum, reach the superficial, molecular layer of the cerebellum. Here, the axons of the cells of the granular layer and the creeping fibers divide in a T-shape, and in the molecular layer their branches take a direction longitudinal to the surface of the cerebellum. The impulses that have reached the molecular layer of the cortex, having passed through the synaptic contacts, fall on the branching dendrites of Purkinje cells located here. Then they follow the dendrites of Purkinje cells to their bodies, located at the border of the molecular and granular layers. Then, along the axons of the same cells crossing the granular layer, they penetrate into the depth of the white matter. The axons of Purkinje cells end in the nuclei of the cerebellum. Mainly in the dentate nucleus. Efferent impulses coming from the cerebellum along the axons of the cells that make up its nucleus and taking part in the formation of the cerebellar peduncles leave the cerebellum.
The cerebellum has three pairs of legs: bottom, middle and top. The lower leg connects it with the medulla oblongata, the middle - with the bridge, the upper - with the midbrain. The legs of the brain make up the pathways that carry impulses to and from the cerebellum.
The cerebellar vermis ensures the stabilization of the body's center of gravity, its balance, stability, regulation of the tone of reciprocal muscle groups, mainly the neck and trunk, and the emergence of physiological cerebellar synergies that stabilize the balance of the body.
To successfully maintain the balance of the body, the cerebellum constantly receives information passing along the spinocerebellar pathways from the proprioceptors of various parts of the body, as well as from the vestibular nuclei, inferior olives, the reticular formation and other formations involved in controlling the position of body parts in space. Most of the afferent pathways leading to the cerebellum pass through the lower cerebellar pedicle, some of them are located in the superior cerebellar pedicle.
Impulses of proprioceptive sensitivity, going to the cerebellum, like other sensory impulses, following the dendrites of the first sensory neurons, reach their bodies located in the spinal nodes. Subsequently, impulses going to the cerebellum along the axons of the same neurons are directed to the bodies of second neurons, which are located in the inner parts of the base of the posterior horns, forming the so-called Clark's pillars. Their axons fall into the lateral sections of the lateral cords of the spinal cord, where they form spinocerebellar pathways, in this case, part of the axons falls into the lateral column of the same side and forms there the posterior spinocerebellar tract of Flexig (tractus spinocerebellaris posterior). Another part of the axons of the cells of the posterior horns passes to the other side of the spinal cord and enters the opposite lateral cord, forming in it anterior spinocerebellar tract of Govers (tractus spinocerebellaris anterior). The spinocerebellar tracts, increasing in volume at the level of each spinal segment, rise to the medulla oblongata.
In the medulla oblongata, the posterior spinocerebellar pathway deviates in the lateral direction and, having passed through the lower cerebellar pedicle, penetrates into the cerebellum. The anterior spinocerebellar pathway passes through the medulla oblongata, the pons of the brain, and reaches the midbrain, at the level of which it makes its second intersection in the anterior cerebral velum and passes into the cerebellum through the superior cerebellar peduncle.
Thus, of the two spinal tracts, one is never crossed (uncrossed Fleksig's path), and the other passes to the opposite side twice (twice crossed by Govers). As a result, both conduct impulses from each half of the body, mainly to the homolateral half of the cerebellum.
In addition to Fleksig's spinocerebellar tracts, impulses to the cerebellum pass through the lower cerebellar pedicle along vestibulocerebellar tract (tractus vestibulocerebellaris), starting mainly in the upper vestibular nucleus of ankylosing spondylitis, and along olivomocerebellar tract (tractus olivocerebellaris) coming from the lower olive. Part of the axons of the cells of the thin and wedge-shaped nuclei, not taking part in the formation of the bulbothalamic tract, in the form of external arcuate fibers (fiber arcuatae externae) also enters the cerebellum through the inferior cerebellar peduncle.
Through its middle legs, the cerebellum receives impulses from the cerebral cortex. These impulses pass through cortical-cerebellopontine pathways, consisting of two neurons. The bodies of the first neurons are located in the cerebral cortex, mainly in the cortex of the posterior parts of the frontal lobes. Their axons pass as part of the radiant crown, the anterior leg of the inner capsule and end in the nuclei of the bridge. Axons of cells of second neurons, whose bodies are located in their own nuclei of the bridge, go to its opposite side and make up, after the intersection, the middle cerebellar pedicle,
ending in the opposite hemisphere of the cerebellum.
Some of the impulses that have arisen in the cerebral cortex reach the opposite hemisphere of the cerebellum, bringing information not about the produced, but only about the planned active movement. Having received such information, the cerebellum instantly sends out impulses that correct voluntary movements, mainly, by extinguishing inertia and the most rational regulation of reciprocal muscle tone - muscle agonists and antagonists. As a result, a kind of eimetry, making voluntary movements clear, perfected, devoid of inappropriate components.
The pathways that leave the cerebellum are composed of the axons of the cells, whose bodies form its nuclei. Most efferent pathways, including pathways from the dentate nuclei, leave the cerebellum through its upper leg. At the level of the lower tubercles of the quadruple, the efferent cerebellar tract crosses (intersection of the upper cerebellar legs of Werneking). After crossing each one of them reaches the red nuclei of the opposite side of the midbrain. In the red nuclei, cerebellar impulses switch to the next neuron and then move along the axons of cells, the bodies of which are embedded in the red nuclei. These axons are formed in red-spinal pathways (tracti rubro spinalis), the paths of Monakov, which soon after exits from red kernels undergo a cross (tire cross or Trout cross), after which they descend into the spinal cord. In the spinal cord, the red-nuclear spinal pathways are located in the lateral cords; their constituent fibers end at the cells of the anterior horns of the spinal cord.
The entire efferent pathway from the cerebellum to the cells of the anterior horns of the spinal cord can be called cerebellar-red-nuclear-spinal (tractus cerebello-rubrospinalis). He crosses twice (intersection of the superior cerebellar peduncles and intersection of the operculum) and ultimately connects each cerebellar hemisphere with peripheral motor neurons located in the anterior horns of the homolateral half of the spinal cord.
From the nuclei of the cerebellar vermis, the efferent pathways go mainly through the lower cerebellar pedicle to the reticular formation of the brain stem and the vestibular nuclei. From here, along the reticulospinal and vestibulospinal pathways passing along the anterior cords of the spinal cord, they also reach the cells of the anterior horns. Part of the impulses coming from the cerebellum, passing through the vestibular nuclei, enters the medial longitudinal bundle, reaches the nuclei III, IV and VI of the cranial nerves that provide the movement of the eyeballs, and affects their function.
In summary, the following should be emphasized:
1. Each half of the cerebellum receives impulses mainly a) from the homolateral half of the body, b) from the opposite hemisphere of the brain, which has cortico-spinal connections with the same half of the body.
(2) From each half of the cerebellum, efferent impulses are directed to the cells of the anterior horns of the homolateral half of the spinal cord and to the nuclei of the cranial nerves that provide movement of the eyeballs.
This nature of the cerebellar connections makes it possible to understand why, when one half of the cerebellum is affected, cerebellar disorders occur mainly in the same, i.e. homolateral, half of the body. This is especially pronounced when the cerebellar hemispheres are affected.
7.2. RESEARCH OF THE FUNCTIONS OF THE CEREBELLA
AND CLINICAL MANIFESTATIONS OF ITS DEFEATS
With damage to the cerebellum, disorders of statics and coordination of movements, muscle hypotonia and nystagmus are characteristic.
Cerebellar lesion first of all his worm, leads to violations of statics - the ability to maintain a stable position of the center of gravity of the human body, balance, stability. When this function is disturbed, static ataxia (from the Greek ataxia - disorder, instability). The instability of the patient is noted. Therefore, in a standing position, he spreads his legs wide apart, balances with his hands. Especially clearly static ataxia is detected with an artificial decrease in the area of support, in particular in the Romberg pose. The patient is invited to stand up, firmly moving his feet and slightly raising his head. In the presence of cerebellar disorders, the patient's instability in this position is noted, his body sways, sometimes he is “pulled” in a certain direction, and if the patient is not supported, he may fall. In the case of damage to the cerebellar worm, the patient usually sways from side to side and often falls back. With pathology of the cerebellar hemisphere, there is a tendency to fall mainly towards the pathological focus. If the static disorder is moderately expressed, it is easier to identify in the so-called complicated or sensitized Romberg pose. The patient is asked to put his feet in one line so that the toe of one foot rests on the heel of the other. The stability assessment is the same as in the usual Romberg position.
Normally, when a person is standing, the muscles of his legs are tense. (support reaction), with the threat of falling to the side, his leg on this side moves in the same direction, and the other leg comes off the floor (jump reaction). With damage to the cerebellum (mainly the worm), the patient's reactions are disturbed
support and jump. Violation of the support reaction is manifested by the patient's instability in the standing position, especially in the Romberg position. Violation of the jump reaction leads to the fact that if the doctor, standing behind the patient and insuring him, pushes the patient in one direction or another, then the patient falls with a slight push (pushing symptom).
With damage to the cerebellum, the patient's gait is usually changed due to the development statolokomotor ataxia. Cerebellar gait in many ways resembles the gait of a drunk person, therefore it is sometimes called the "gait of a drunk." Due to instability, the patient walks uncertainly, spreading his legs wide apart, while he is "thrown" from side to side. And when the cerebellar hemisphere is damaged, it deviates when walking from a given direction towards the pathological focus. Instability is especially pronounced when cornering. If the ataxia is pronounced, then the patients completely lose the ability to control their body and can not only stand and walk, but even sit.
The predominant lesion of the cerebellar hemispheres leads to a disorder of its anti-inertial effects, in particular to the emergence kinetic ataxia. It is manifested by the awkwardness of movements and is especially pronounced with movements that require precision. To detect kinetic ataxia, tests for coordination of movements are performed. Some of them are described below.
Test for diadochokinesis (from the Greek diadochos - sequence). The patient is invited to close his eyes, stretch his arms forward and quickly, rhythmically supinate and penetrate the hands. In case of damage to the cerebellar hemisphere, the movements of the hand on the side of the pathological process turn out to be more sweeping (a consequence of dysmetria, more precisely, hypermetria), as a result, the hand begins to lag behind. This indicates the presence of adiadochokinesis.
Finger test. A patient with closed eyes should withdraw his hand, and then, slowly, with his index finger, touch the tip of the nose. In the case of cerebellar pathology, the hand on the side of the pathological focus makes an excessive movement in volume (hypermetry), as a result of which the patient misses. A finger-nose test reveals a characteristic of cerebellar pathology cerebellar (intentional) tremor, the amplitude of which increases as the finger approaches the target. This test allows you to identify the so-called bradytelekinesia. (bridle symptom): not far from the target, the movement of the finger slows down, sometimes even pauses, and then resumes again.
Finger-finger test. A patient with closed eyes is invited to spread his hands wide and then bring the index fingers closer, trying to get the finger into the finger, while, as with the finger test, an intentional tremor and a bridle symptom are revealed.
Calcaneal knee test (fig. 7.3). The patient, lying on his back with closed eyes, is asked to raise one leg high and then hit the knee of the other leg with its heel. With cerebellar pathology, the patient cannot or it is difficult for him to get his heel into the knee of the other leg, especially when performing a test with the leg homolateral to the affected cerebellar hemisphere. If, nevertheless, the heel reaches the knee, then it is proposed to hold it, slightly touching the front surface of the lower leg, down to the ankle joint, while in the case of cerebellar pathology, the heel slides off the lower leg all the time in one direction or the other.

Rice. 7.3.Calcaneal knee test.
Indicative test: The patient is invited to hit the rubber tip of the hammer, which is in the examiner's hand, with his index finger several times. In the case of cerebellar pathology in the patient's hand on the side of the affected cerebellar hemisphere, there is a misalignment due to dysmetria.
Tom-Jumenti symptom: If the patient picks up an object, such as a glass, he spreads his fingers excessively.
Cerebellar nystagmus. Twitching of the eyeballs when looking to the sides (horizontal nystagmus) is considered as a consequence of intentional tremor of the eyeballs (see chapter 30).
Speech disorder: Speech loses its fluidity, becomes explosive, fragmented, chanted like cerebellar dysarthria (see Chapter 25).
Changing handwriting: Due to the disorder in the coordination of hand movements, the handwriting becomes uneven, the letters are deformed, excessively large (megalography).
Pronatory phenomenon: The patient is asked to keep his arms outstretched in the supination position, while spontaneous pronation soon occurs on the side of the affected cerebellar hemisphere.
Hoffa-Schilder symptom: If the patient holds his arms outstretched forward, then on the side of the affected hemisphere, the arm is soon withdrawn outward.
An imitation phenomenon. A patient with closed eyes should quickly give the hand a position similar to that which the examiner had previously given to his other hand. When the cerebellar hemisphere is damaged, the homolateral hand makes a movement that is excessive in amplitude.
Doinikov's phenomenon. Finger phenomenon. The seated patient is invited to put supinated hands with fingers apart on his thighs and close his eyes. In the case of a lesion of the cerebellum on the side of the pathological focus, spontaneous flexion of the fingers and pronation of the hand and forearm soon occur.
Stuart-Holmes symptom. The examiner asks the patient sitting on the chair to bend the supinated forearms and at the same time, taking his hands by the wrists, resists him. If at the same time you suddenly release the patient's hands, then the hand on the affected side, bending by inertia, will forcefully hit him in the chest.
Muscle hypotension. The defeat of the cerebellar vermis usually leads to diffuse muscle hypotension. With the defeat of the cerebellar hemisphere, passive movements reveal a decrease in muscle tone on the side of the pathological process. Muscle hypotonia leads to the possibility of hyperextension of the forearm and lower leg (Olshansky symptom) with passive movements, to the appearance symptoms of a dangling hand or foot with their passive shaking.
Pathological cerebellar asynergies. Violations of physiological synergies during complex motor acts are revealed, in particular, during the following tests (Fig. 7.4).
1. Asynergy according to Babinsky in a standing position. If a patient standing with shifted legs tries to bend back, throwing his head back, then normally in this case, flexion of the knee joints occurs. In cerebellar pathology due to asynergy, this friendly movement is absent, and the patient, losing balance, falls back.
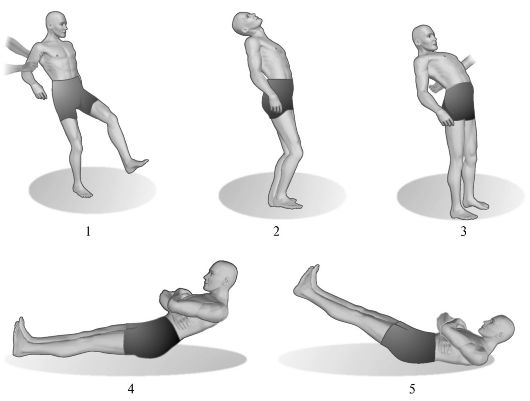
Rice. 7.4.Cerebellar asynergy.
1 - gait of a patient with severe cerebellar ataxia; 2 - back tilt of the body is normal; 3 - with damage to the cerebellum, the patient, bending backward, cannot maintain balance; 4 - performing a test for cerebellar asynergy according to Babinsky by a healthy person; 5 - performing the same test in patients with cerebellar lesions.
2. Asynergy according to Babinsky in the supine position. The patient, lying on a firm plane with legs extended, spread to the width of the shoulder girdle, is invited to cross his arms over his chest and then sit down. In the presence of cerebellar pathology due to the absence of friendly contraction of the gluteal muscles (manifestation of asynergy), the patient cannot fix the legs and pelvis on the support area, as a result, the legs rise and he cannot sit down. The significance of this symptom should not be overestimated in elderly patients, in people with a flabby or obese abdominal wall.
Summarizing the above, the diversity and importance of the functions performed by the cerebellum should be emphasized. As part of a complex feedback regulatory mechanism, the cerebellum acts as a focal point to balance the body and maintain muscle tone. As P. Duus (1995) notes, the cerebellum provides the ability to perform discrete and precise movements, the author reasonably believes that the cerebellum works like a computer, tracking and coordinating sensory information at the input and simulating motor signals at the output.
7.3. MULTISYSTEM DEGENERATIONS
With signs of cerebellar pathology
Multisystem degenerations are a group of neurodegenerative diseases, the common feature of which is the multifocal nature of the lesion with the involvement of various functional and neurotransmitter systems of the brain in the pathological process and, therefore, the polysystemic nature of clinical manifestations.
7.3.1. Cerebellar ataxia
Spinocerebellar ataxias include progressive hereditary degenerative diseases, in which the structures of the cerebellum, the brain stem and the pathways of the spinal cord, which are mainly related to the extrapyramidal system, are mainly affected.
7.3.1.1. Friedreich's hereditary ataxia
Hereditary disease described in 1861 by the German neuropathologist N. Friedreich (Friedreich N., 1825-1882). It is inherited in an autosomal recessive manner or (less often) in an autosomal dominant manner with incomplete penetrance and variable gene expression. Sporadic cases of the disease are also possible.
Pathogenesisdisease not specified. In particular, there is no idea about the primary biochemical defect that constitutes its basis.
Pathomorphology.Pathological studies reveal a pronounced thinning of the spinal cord due to atrophic processes in its posterior and lateral cords. As a rule, the wedge-shaped (Burdach) and gentle (Gaulle) pathways and spinocerebellar pathways of Govers and Fleksig suffer, as well as the crossed pyramidal pathway containing
many fibers related to the extrapyramidal system. Degenerative processes are also expressed in the cerebellum, in its white matter and in the nuclear apparatus.
Clinical manifestations. The disease manifests itself in children or young people under the age of 25. S.N. Davidenkov (1880-1961) noted that more often the clinical signs of the disease occur in children 6-10 years of age. The first sign of illness is usually ataxia. Patients experience uncertainty, staggering when walking, gait changes (when walking, legs are wide apart). The gait in Friedreich's disease can be called tabetic-cerebellar, since its changes are caused by a combination of sensitive and cerebellar ataxia, as well as a usually pronounced decrease in muscle tone. Disorders of statics, discoordination in the hands, intentional tremor, dysarthria are also characteristic. Possible nystagmus, hearing loss, elements of chanting speech, signs of pyramidal insufficiency (tendon hyperreflexia, foot pathological reflexes, sometimes a slight increase in muscle tone), imperative urge to urinate, decreased sexual potency. Sometimes athetoid hyperkinesis appears.
An early-onset disorder of deep sensitivity leads to a progressive decrease in tendon reflexes: first on the legs, and then on the arms. Over time, muscle hypotrophy of the distal legs forms. The presence of anomalies in the development of the skeleton is characteristic. First of all, this is manifested by the presence Friedreich's feet: the foot is shortened, "hollow", with a very high arch. The main phalanges of her fingers are unbent, the rest are bent (Fig. 7.5). Possible deformation of the spine, chest. There are often manifestations of cardiopathy. The disease progresses slowly, but steadily leads to the disability of patients who eventually become bedridden.
Treatment. Pathogenetic treatment has not been developed. Prescribe drugs that improve metabolism in the structures of the nervous system, fortifying agents. With severe deformity of the feet, orthopedic shoes are indicated.
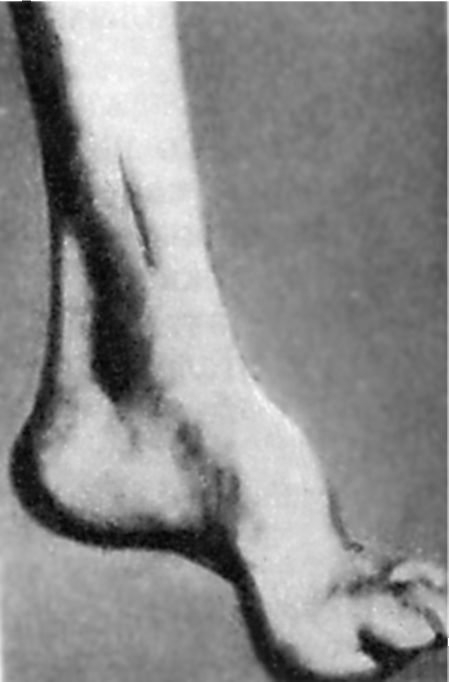
Rice. 7.5.Friedreich's foot.
7.3.1.2. Hereditary cerebellar ataxia (Pierre Marie's disease)
This is a chronic progressive hereditary disease, manifested at the age of 30-45 years, with slowly growing cerebellar disorders in combination with signs of pyramidal insufficiency, while static and dynamic cerebellar ataxia, intentional tremors, chanted speech, tendon hyperreflexia are characteristic. Possible clonuses, pathological pyramidal reflexes, strabismus, decreased vision, narrowing of the visual fields due to primary atrophy of the optic nerves and pigmentary degeneration of the retina. The course of the disease is slowly progressive. There is a decrease in the size of the cerebellum, cell degeneration
Purkinje, inferior olives, spinocerebellar tracts. It is inherited in an autosomal dominant manner. The disease was described in 1893 by the French neuropathologist R. Marie (1853-1940).
Currently, there is no unanimity in the understanding of the term "Pierre Marie's disease", and the question of the possibility of separating it into an independent nosological form is debatable.
No treatment has been developed. Usually, metabolically active and restorative, as well as symptomatic agents are used.
7.3.2. Olivopontocerebellar dystrophy (Dejerine-Thom disease)
This is a group of chronic progressive hereditary diseases, in which dystrophic changes develop mainly in the cerebellum, lower olives, in the pons' own nuclei and in the brain structures associated with them.
With the development of the disease at a young age, about half of the cases are inherited in a dominant or recessive manner, the rest are sporadic. In sporadic cases of the disease, manifestations of akinetic-rigid syndrome and progressive autonomic failure are more common. The average age of the patient with the manifestation of the hereditary form of the disease in the phenotype is 28 years, with the sporadic - 49 years, the average life expectancy is 14.9 and 6.3 years, respectively. In the sporadic form, in addition to atrophy of the olives, pons and cerebellum, lesions of the lateral cords of the spinal cord, substantia nigra and striatum, a bluish spot in the rhomboid fossa of the fourth ventricle of the brain are more often found.
Symptoms of the growing cerebellar syndrome are characteristic. Disorders of sensitivity, elements of bulbar and akinetic-rigid syndromes, hyperkinesis, in particular myorrhythmias in the tongue and soft palate, ophthalmoparesis, decreased visual acuity, intellectual disorders are possible. The disease was described in 1900 by the French neuropathologists J. Dejerine and A. Thomas.
The disease often debuts with disturbances in walking - instability, discoordination, unexpected falls are possible. These disorders may be the only manifestation of the disease within 1-2 years. In the future, coordination disorders in the hands arise and grow: manipulations with small objects are difficult, handwriting is disturbed, an intentional tremor occurs. Speech becomes intermittent, blurry, with a nasal tinge and the rhythm of breathing that does not correspond to the structure of speech (the patient speaks as if he is being strangled). At this stage of the disease, manifestations of progressive autonomic insufficiency join, signs of akinetic-rigid syndrome appear. Sometimes the dominant symptoms for the patient are dysphagia, attacks of nocturnal suffocation. They develop in connection with mixed paresis of the bulbar muscles and can be life-threatening.
In 1970, the German neuropathologists B.W. Konigsmark and L.P. Weiner singled out 5 main types olivopontocerebellar dystrophy, differing either in clinical and morphological manifestations, or in the type of inheritance.
I type (Menzel type). At the age of 14-70 (more often 30-40) years, it manifests itself ataxia, dysarthria, dysphonia, muscle hypotonia, in the late stage - a gross tremor of the head, trunk, arms, muscles, signs of akinetic-rigid syndrome. Possible pathological pyramidal signs, gaze paresis, external and internal ophthalmoplegia, sensitivity disorders, dementia. It is inherited in an autosomal dominant manner. It was singled out as an independent form in 1891 by P. Menzel.
II type (Fickler-Winkler type) ... At the age of 20-80, it manifests itself ataxia, decreased muscle tone and tendon reflexes. It is inherited in an autosomal recessive manner. Sporadic cases are possible.
III type with retinal degeneration. It manifests itself in childhood or young (up to 35 years) age ataxia, tremor of the head and extremities, dysarthria, signs of pyramidal insufficiency, progressive decrease in vision with an outcome in blindness; possible nystagmus, ophthalmoplegia, sometimes dissociated sensory disorders. It is inherited in an autosomal dominant manner.
IV type (Jester-Highmaker type). At the age of 17-30 years, he debuts with cerebellar ataxia or signs of lower spastic paraparesis, in both cases, already at an early stage of the disease, a combination of these manifestations is formed, to which elements of bulbar syndrome, paresis of facial muscles, and deep sensitivity disorders are subsequently added. Dominant inherited.
V type of. It manifests itself at the age of 7-45 years ataxia, dysarthria, signs of akinetic-rigid syndrome and other extrapyramidal disorders, progressive ophthalmoplegia and dementia are possible. Dominant inherited.
7.3.3. Olivorubrocerebellar degeneration (Lejeune-Lermitte syndrome, Lermitte disease)
The disease is characterized by progressive atrophy of the cerebellum, mainly of its cortex, dentate nuclei and upper cerebellar peduncles, inferior olives, and red nuclei. It is manifested primarily by static and dynamic ataxia; in the future, other signs of cerebellar syndrome and damage to the brain stem are possible. The disease was described by the French neuropathologists J. Lhermitte (Lhermitte J.J., 1877-1959) and J. Lezhon (Lejonne J., born in 1894).
7.3.4. Multisystem atrophy
In recent decades, a sporadic, progressive neurodegenerative disease called multisystem atrophy has been identified as an independent form. It is characterized by a combined lesion of the basal ganglia, cerebellum, brain stem, spinal cord. The main clinical manifestations: parkinsonism, cerebellar ataxia, signs of pyramidal and autonomic insufficiency (Levin O.S., 2002). Depending on the predominance of certain features of the clinical picture, three types of multisystem atrophy are distinguished.
1) olivopontocerebellar type, characterized by the predominance of signs of cerebellar attack;
2) strionigral type, in which signs of parkinsonism dominate;
3) Shai-Drager syndrome, characterized by the predominance in the clinical picture of signs of progressive autonomic failure with symptoms of orthostatic arterial hypotension.
The basis of multisystem atrophy is the selective degeneration of certain areas of the predominantly gray matter of the brain with damage to neurons and glial elements. The causes of degenerative manifestations in the brain tissue remain unknown today. The manifestations of multisystem atrophy of the olivopontocerebellar type are associated with damage to Purkinje cells in the cerebellar cortex, as well as neurons of the inferior olives, pontocerebellar nuclei, demyelination and degeneration, mainly of the pontocerebellar pathways.
Cerebellar disorders are usually static and dynamic ataxia with impaired locomotor movement. Characterized by instability in the Romberg position, ataxia when walking, dysmetria, adiadochokinesis, intentional tremor, there may be nystagmus (horizontal vertical, beating down), intermittent and slow tracking gaze movements, impaired convergence of the eyes, chanted speech.
Multisystem atrophy usually occurs in adulthood and progresses rapidly. Diagnosis is based on clinical evidence and is characterized by a combination of signs of parkinsonism, cerebellar failure, and autonomic disorders. Treatment of the disease has not been developed. The duration of the disease - within 10 years, ends in death.
7.4. OTHER DISEASES ASSOCIATED WITH SYMPTOMS OF CEREBRAL DISEASE
If the patient shows signs of cerebellar lesion, then in most cases, first of all you have to think about the possibility cerebellar tumors(astrocytoma, angioblastoma, medulloblastoma, metastatic tumors) or multiple sclerosis. At cerebellar tumors early signs of intracranial hypertension appear. In multiple sclerosis, it is usually possible to identify, in addition to cerebellar pathology, clinical manifestations of damage to other structures of the central nervous system, primarily the visual and pyramidal systems. In classical neurology, the characteristic of multiple sclerosis Charcot's triad: nystagmus, intentional tremor and chanted speech, and Nonne's syndrome: disorder of coordination of movements, dysmetria, chanted speech and cerebellar asynergy.
Cerebellar disorders are major and in post-traumatic Mann syndrome, which is characterized by ataxia, discoordination, asynergy, nystagmus. Trauma or infection can cause cerebellar Goldstein-Reichmann syndrome: disorders of statics and coordination of movements, asynergy, intentional tremor, decreased muscle tone, hypermetria, megalography, impaired perception of the mass (weight) of an object in the hands.
Disorders of cerebellar function can also be congenital in nature, manifesting, in particular, Zeeman's syndrome: ataxia, delayed speech development, and subsequently cerebellar dysarthria.
Congenital cerebellar ataxia It is manifested by a delay in the development of the child's motor functions (at the age of 6 months he cannot sit, he begins to walk late, while the gait is atactic), as well as delayed speech, prolonged preservation of dysarthria, sometimes lagging behind in mental development, and microcranial manifestations are not uncommon. On CT, the cerebellar hemispheres are reduced. By about 10 years of age, compensation of brain functions usually occurs, which, however, can be disrupted under the influence of harmful exogenous influences. Progressive forms of the disease are also possible.
A manifestation of congenital hypoplasia of the cerebellum is and Fancony-Turner syndrome. It is characterized by impaired statics and coordination of movements, nystagmus, which are usually accompanied by mental retardation.
Congenital also includes an autosomal recessive inherited type, which is rarely found Betten's disease: It is characterized by congenital cerebellar ataxia, manifested in the first year of life by impaired statics and coordination of movements, nystagmus, gaze coordination disorder, and moderate muscle hypotonia. Dysplastic signs are possible. The child is late, sometimes only at 2-3 years of age, begins to hold his head, even later - to stand, walk, talk. His speech is changed according to the type of cerebellar dysarthria. Vegetative-visceral disorders, manifestations of immunosuppression are possible. After a few years, the clinical picture usually stabilizes, the patient to some extent adapts to the existing defects.
Spastic ataxia at the suggestion of A. Bell and E. Carmichel (1939), cerebellar ataxia inherited by an autosomal dominant type was named, which is characterized by the onset of the disease at 3-4 years of age and is manifested by a combination of cerebellar ataxia with dysarthria, tendon hyperreflexia and increased muscle tone by spastic type, while possible (but not obligate signs of the disease) atrophy of the optic nerves, retinal degeneration, nystagmus, oculomotor disorders.
Autosomal dominant is inherited Feldman syndrome(described by the German physician H. Feldmann, born in 1919): cerebellar ataxia, intentional tremors and early graying of hair. It manifests itself in the second decade of life and further slowly progresses, leading to disability after 20-30 years.
Late cerebellar atrophy or Tom's syndrome described in 1906 by the French neurologist A. Thomas (1867-1963), usually manifests itself in persons over 50 years of age with progressive atrophy of the cerebellar cortex. In the phenotype, signs of cerebellar syndrome appear, primarily cerebellar static and locomotor ataxia, chanted speech, and changes in handwriting. In a far advanced stage, manifestations of pyramidal insufficiency are possible.
The combination of cerebellar disorders with myoclonus is characterized by Hunt myoclonic cerebellar dyssynergia, or myoclonus ataxia, with this symptom complex in the clinical picture, intentional tremor, myoclonus arising in the hands, and subsequently acquiring a generalized character, ataxia and dyssynergia, nystagmus, chanted speech, decreased muscle tone are manifested. It is a consequence of degeneration of the cerebellar nuclei, red nuclei and their connections, as well as cortical-subcortical structures.
In an advanced stage of the disease, epileptic seizures and dementia are possible. The prognosis is bad. Refers to a rare form of progressive hereditary ataxia. It is inherited in an autosomal recessive manner. It usually appears at a young age. The nosological independence of the symptom complex is disputed. The American neurologist R. Hunt (1872-1937) described the disease in 1921.
Among degenerative processes, a certain place is occupied by Holmes cerebellar degeneration, or familial cerebellar atrophy, or progressive atrophy of the cerebellar system, mainly of the dentate nuclei, as well as red nuclei, while manifestations of demyelination are expressed in the superior cerebellar pedicle. Characterized by static and dynamic ataxia, asynergia, nystagmus, dysarthria, decreased muscle tone, muscular dystonia, head tremor, myoclonus. Epileptic seizures appear almost simultaneously. Intelligence is usually preserved. EEG shows paroxysmal dysrhythmia. The disease is recognized as hereditary, but the type of inheritance is not specified. Described the disease in 1907 by the English neuropathologist G. Holmes
(1876-1965).
Alcoholic cerebellar degeneration - a consequence of chronic alcohol intoxication. Mainly the cerebellar worm is affected, with cerebellar ataxia and impaired coordination of leg movements primarily manifested, while hand movements, oculomotor and speech functions are impaired to a much lesser extent. Usually this disease is accompanied by a pronounced memory loss in combination with polyneuropathy.
manifests itself as cerebellar ataxia, which can sometimes be the only clinical symptom associated with a malignant tumor, without local signs indicating the place of its occurrence. Paraneoplastic cerebellar degeneration may be, in particular, a secondary manifestation of breast or ovarian cancer.
Barraquer-Bordas-Ruiz-Lara Syndrome manifests itself as cerebellar disorders arising in connection with rapidly progressive atrophy of the cerebellum. A syndrome in patients with bronchial cancer accompanied by general intoxication is described by the modern Spanish physician L. Barraquer-Bordas (born in 1923).
Rarely found recessive X chromosomal ataxia- a hereditary disease that manifests itself almost only in men with slowly progressive cerebellar insufficiency. It is transmitted in a recessive, sex-linked type.
Noteworthy and familial paroxysmal ataxia, or periodic ataxia. It makes its debut more often in childhood, but it can also appear later - up to 60 years. The clinical picture is reduced to paroxysmal manifestations of nystagmus, dysarthria and ataxia, decreased muscle tone, dizziness, nausea, vomiting, headache, lasting from several minutes to 4 weeks.
Attacks of familial paroxysmal ataxia can be triggered by emotional stress, physical fatigue, fever, alcohol intake, while between attacks focal neurological symptoms in most cases are not detected, but sometimes nystagmus and mild cerebellar symptoms are possible.
The morphological substrate of the disease is recognized as an atrophic process mainly in the anterior part of the cerebellar worm. The disease was first described in 1946 by M. Parker. It is inherited in an autosomal dominant manner. In 1987, with familial paroxysmal ataxia, a decrease in the activity of pyruvate dehydrogenase of blood leukocytes to 50-60% of the normal level was found. In 1977 R. Lafrance et al. drew attention to the high prophylactic effect of diacarb, later flunarizine was proposed for the treatment of familial paroxysmal ataxia.
Acute cerebellar ataxia or Leiden-Westphal syndrome, is a well-defined symptom complex, which is a parainfectious complication. It occurs more often in children 1-2 weeks after a general infection (influenza, typhus, salmonellosis, etc.). Characterized by gross static and dynamic ataxia, intentional tremor, hypermetria, asynergia, nystagmus, chanted speech, decreased muscle tone. In the cerebrospinal fluid, lymphocytic pleocytosis, a moderate increase in protein, is detected. At the onset of the disease, dizziness, disturbances of consciousness, convulsions are possible. On CT and MRI, pathology is not detected. The course is benign. In most cases, after a few weeks or months - complete recovery, sometimes - residual disorders in the form of mild cerebellar insufficiency.
Marie-Foix-Alajuanin's disease - late symmetric cortical atrophy of the cerebellum with a predominant lesion of piriform neurons (Purkinje cells) and the granular layer of the cortex, as well as the oral part of the cerebellar vermis and olive degeneration. It manifests itself in persons of 40-75 years of age with balance disorder, ataxia, gait disturbance, coordination disorders and decreased muscle tone, mainly in the legs; the intentional tremor in the hands is not very pronounced. Speech disorders are possible, but do not belong to the obligate signs of the disease. The disease was described in 1922 by the French neuropathologists P. Marie, Ch. Foix and Th. Alajouanine. The disease is sporadic. The etiology of the disease has not been clarified. There are opinions about the provoking role of intoxication, primarily alcohol abuse, as well as hypoxia, hereditary burden. The clinical picture is confirmed by CT data of the head, which reveals a pronounced decrease in the volume of the cerebellum against the background of diffuse atrophic processes in the brain. In addition, a high level of aminotransferases in blood plasma is recognized as characteristic (Ponomareva E.N. et al., 1997).
